Naomi Rapier-Sharman, Jeffrey Clancy, Brett E Pickett
{"title":"非霍奇金 B 细胞淋巴瘤的联合二级转录组分析预测了与细胞外基质相关的途径和可靠的诊断生物标志物。","authors":"Naomi Rapier-Sharman, Jeffrey Clancy, Brett E Pickett","doi":"10.26502/jbsb.5107040","DOIUrl":null,"url":null,"abstract":"<p><p>Approximately 450,000 cases of Non-Hodgkin's lymphoma are annually diagnosed worldwide, resulting in ~240,000 deaths. An augmented understanding of the common mechanisms of pathology among larger numbers of B-cell Non-Hodgkin's Lymphoma (BCNHL) patients is sorely needed. We consequently performed a large joint secondary transcriptomic analysis of the available BCNHL RNA-sequencing projects from GEO, consisting of 322 relevant samples across ten distinct public studies, to find common underlying mechanisms and biomarkers across multiple BCNHL subtypes and patient subpopulations; limitations may include lack of diversity in certain ethnicities and age groups and limited clinical subtype diversity due to sample availability. We found ~10,400 significant differentially expressed genes (FDR-adjusted p-value < 0.05) and 33 significantly modulated pathways (Bonferroni-adjusted p-value < 0.05) when comparing BCNHL samples to non-diseased B-cell samples. Our findings included a significant class of proteoglycans not previously associated with lymphomas as well as significant modulation of genes that code for extracellular matrix-associated proteins. Our drug repurposing analysis predicted new candidates for repurposed drugs including ocriplasmin and collagenase. We also used a machine learning approach to identify robust BCNHL biomarkers that include YES1, FERMT2, and FAM98B, which have not previously been associated with BCNHL in the literature, but together provide ~99.9% combined specificity and sensitivity for differentiating lymphoma cells from healthy B-cells based on measurement of transcript expression levels in B-cells. This analysis supports past findings and validates existing knowledge while providing novel insights into the inner workings and mechanisms of transformed B-cell lymphomas that could give rise to improved diagnostics and/or therapeutics.</p>","PeriodicalId":73617,"journal":{"name":"Journal of bioinformatics and systems biology : Open access","volume":"5 4","pages":"119-135"},"PeriodicalIF":0.0000,"publicationDate":"2022-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9980876/pdf/","citationCount":"0","resultStr":"{\"title\":\"Joint Secondary Transcriptomic Analysis of Non-Hodgkin's B-Cell Lymphomas Predicts Reliance on Pathways Associated with the Extracellular Matrix and Robust Diagnostic Biomarkers.\",\"authors\":\"Naomi Rapier-Sharman, Jeffrey Clancy, Brett E Pickett\",\"doi\":\"10.26502/jbsb.5107040\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Approximately 450,000 cases of Non-Hodgkin's lymphoma are annually diagnosed worldwide, resulting in ~240,000 deaths. An augmented understanding of the common mechanisms of pathology among larger numbers of B-cell Non-Hodgkin's Lymphoma (BCNHL) patients is sorely needed. We consequently performed a large joint secondary transcriptomic analysis of the available BCNHL RNA-sequencing projects from GEO, consisting of 322 relevant samples across ten distinct public studies, to find common underlying mechanisms and biomarkers across multiple BCNHL subtypes and patient subpopulations; limitations may include lack of diversity in certain ethnicities and age groups and limited clinical subtype diversity due to sample availability. We found ~10,400 significant differentially expressed genes (FDR-adjusted p-value < 0.05) and 33 significantly modulated pathways (Bonferroni-adjusted p-value < 0.05) when comparing BCNHL samples to non-diseased B-cell samples. Our findings included a significant class of proteoglycans not previously associated with lymphomas as well as significant modulation of genes that code for extracellular matrix-associated proteins. Our drug repurposing analysis predicted new candidates for repurposed drugs including ocriplasmin and collagenase. We also used a machine learning approach to identify robust BCNHL biomarkers that include YES1, FERMT2, and FAM98B, which have not previously been associated with BCNHL in the literature, but together provide ~99.9% combined specificity and sensitivity for differentiating lymphoma cells from healthy B-cells based on measurement of transcript expression levels in B-cells. This analysis supports past findings and validates existing knowledge while providing novel insights into the inner workings and mechanisms of transformed B-cell lymphomas that could give rise to improved diagnostics and/or therapeutics.</p>\",\"PeriodicalId\":73617,\"journal\":{\"name\":\"Journal of bioinformatics and systems biology : Open access\",\"volume\":\"5 4\",\"pages\":\"119-135\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2022-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9980876/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of bioinformatics and systems biology : Open access\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.26502/jbsb.5107040\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2022/9/27 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of bioinformatics and systems biology : Open access","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.26502/jbsb.5107040","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/9/27 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
摘要
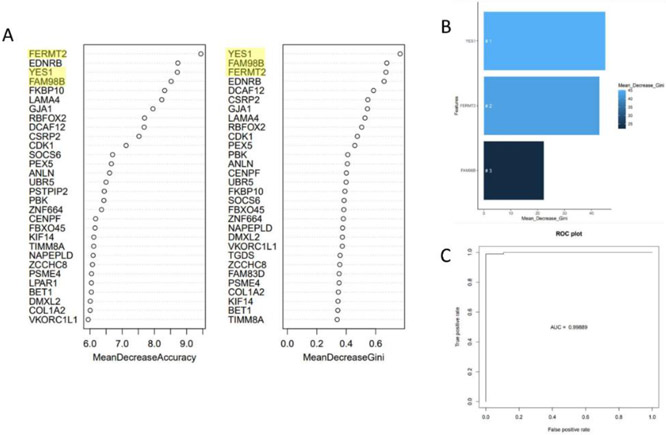
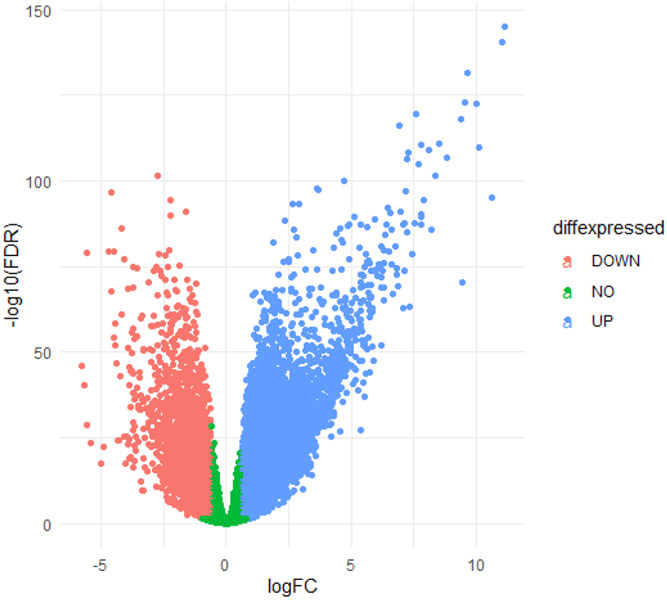
全世界每年诊断出约 450,000 例非霍奇金淋巴瘤,导致约 240,000 人死亡。我们亟需进一步了解更多 B 细胞非霍奇金淋巴瘤(BCNHL)患者的共同病理机制。因此,我们对GEO中现有的BCNHL RNA测序项目进行了大规模的联合二次转录组分析,其中包括十项不同的公开研究中的322个相关样本,目的是在多种BCNHL亚型和患者亚群中寻找共同的潜在机制和生物标志物;局限性可能包括某些种族和年龄组缺乏多样性,以及由于样本可用性而导致临床亚型多样性有限。在将 BCNHL 样本与未患病的 B 细胞样本进行比较时,我们发现了约 10,400 个显著差异表达的基因(经 FDR 调整的 p 值小于 0.05)和 33 个显著调节的通路(经 Bonferroni-adjusted 调整的 p 值小于 0.05)。我们的发现包括一类以前与淋巴瘤无关的重要蛋白多糖,以及编码细胞外基质相关蛋白的基因的重大调控。我们的药物再利用分析预测了新的候选再利用药物,包括奥克立普酶和胶原酶。我们还使用机器学习方法确定了稳健的 BCNHL 生物标记物,其中包括 YES1、FERMT2 和 FAM98B,这些标记物以前在文献中与 BCNHL 无关,但根据 B 细胞中转录物表达水平的测量,它们在区分淋巴瘤细胞和健康 B 细胞方面具有约 99.9% 的综合特异性和敏感性。这项分析支持了过去的研究结果,验证了现有的知识,同时为了解转化B细胞淋巴瘤的内部运作和机制提供了新的视角,有助于改进诊断和/或治疗方法。
Joint Secondary Transcriptomic Analysis of Non-Hodgkin's B-Cell Lymphomas Predicts Reliance on Pathways Associated with the Extracellular Matrix and Robust Diagnostic Biomarkers.
Approximately 450,000 cases of Non-Hodgkin's lymphoma are annually diagnosed worldwide, resulting in ~240,000 deaths. An augmented understanding of the common mechanisms of pathology among larger numbers of B-cell Non-Hodgkin's Lymphoma (BCNHL) patients is sorely needed. We consequently performed a large joint secondary transcriptomic analysis of the available BCNHL RNA-sequencing projects from GEO, consisting of 322 relevant samples across ten distinct public studies, to find common underlying mechanisms and biomarkers across multiple BCNHL subtypes and patient subpopulations; limitations may include lack of diversity in certain ethnicities and age groups and limited clinical subtype diversity due to sample availability. We found ~10,400 significant differentially expressed genes (FDR-adjusted p-value < 0.05) and 33 significantly modulated pathways (Bonferroni-adjusted p-value < 0.05) when comparing BCNHL samples to non-diseased B-cell samples. Our findings included a significant class of proteoglycans not previously associated with lymphomas as well as significant modulation of genes that code for extracellular matrix-associated proteins. Our drug repurposing analysis predicted new candidates for repurposed drugs including ocriplasmin and collagenase. We also used a machine learning approach to identify robust BCNHL biomarkers that include YES1, FERMT2, and FAM98B, which have not previously been associated with BCNHL in the literature, but together provide ~99.9% combined specificity and sensitivity for differentiating lymphoma cells from healthy B-cells based on measurement of transcript expression levels in B-cells. This analysis supports past findings and validates existing knowledge while providing novel insights into the inner workings and mechanisms of transformed B-cell lymphomas that could give rise to improved diagnostics and/or therapeutics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


