A Sarajlija, L Armengol, A Maver, I Kitic, D Prokic, M Cehic, M S Djuricic, B Peterlin
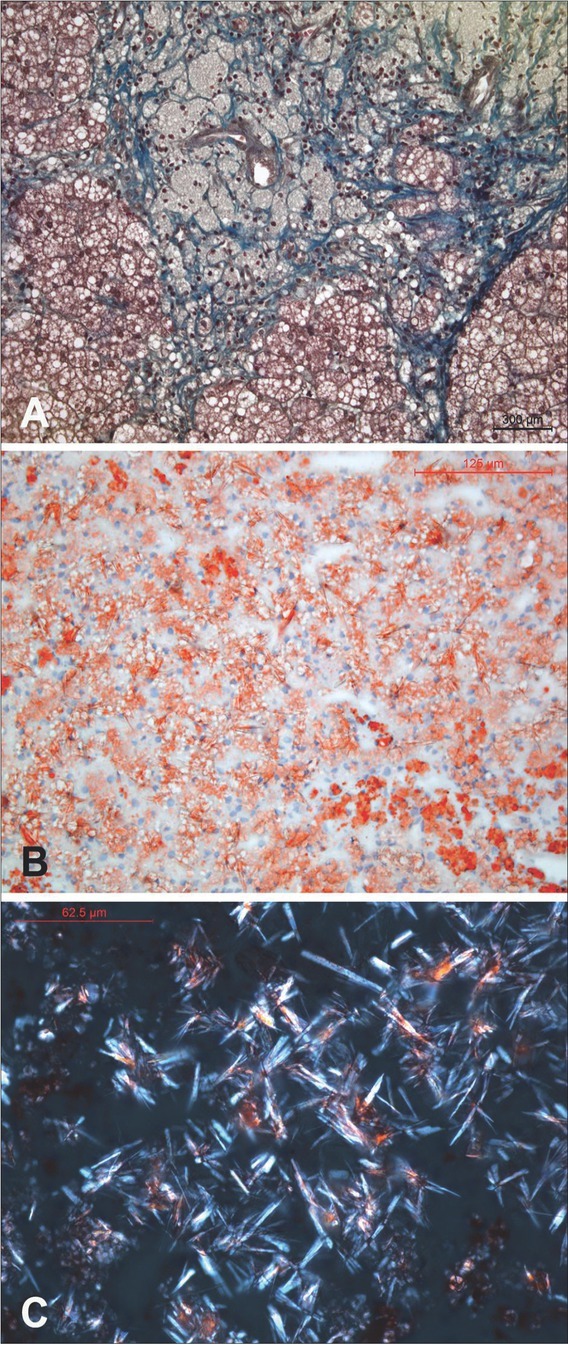
{"title":"与不同表型相关的LIPA基因新变异。","authors":"A Sarajlija, L Armengol, A Maver, I Kitic, D Prokic, M Cehic, M S Djuricic, B Peterlin","doi":"10.2478/bjmg-2022-0010","DOIUrl":null,"url":null,"abstract":"<p><p>Deficiency of lysosomal acid lipase (LAL-D) is caused by biallelic pathogenic variants in the <i>LIPA</i> gene. Spectrum of LAL-D ranges from early onset of hepatosplenomegaly and psychomotor regression (Wolman disease) to a more chronic course (cholesteryl ester storage disease - CESD). The diagnosis is based on lipid and biomarker profiles, specific liver histopathology, enzyme deficiency, and identification of causative genetic variants. Biomarker findings are a useful for diagnostics of LAL-D, including high plasma concentration of chitotriosidase as well as elevated oxysterols. Current treatment options include enzyme replacement therapy (sebelipase-alpha), statins, liver transplantation, and stem cell transplantation. We present two pairs of siblings from Serbia with a distinctive phenotype resembling LAL-D with a novel variant of unknown significance (VUS) detected in the <i>LIPA</i> gene and residual LAL activity. All patients presented with hepatosplenomegaly at early childhood. In siblings from family 1, compound heterozygosity for a pathogenic c.419G>A (p.Trp140Ter) variant and a novel VUS c.851C>T (p.Ser284Phe) was detected. Patients from family 2 were homozygous for c.851C>T VUS and both have typical histopathologic findings for LAL-D in the liver. Enzyme activity of LAL was tested in three patients and reported as sufficient, and therefore enzyme replacement therapy could not be approved. When confronted with a challenge of diagnosing an inherited metabolic disorder, several aspects are taken into consideration: clinical manifestations, specific biomarkers, enzyme assay results, and molecular genetic findings. This report brings cases to light which have a considerable discrepancy between those aspects, namely the preserved LAL enzyme activity in presence of clinical manifestations and rare variants in the <i>LIPA</i> gene.</p>","PeriodicalId":55403,"journal":{"name":"Balkan Journal of Medical Genetics","volume":"25 1","pages":"93-100"},"PeriodicalIF":0.9000,"publicationDate":"2022-06-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/0d/f0/bjmg-25-093.PMC9985358.pdf","citationCount":"0","resultStr":"{\"title\":\"A Novel Variant in the <i>LIPA</i> Gene Associated with Distinct Phenotype.\",\"authors\":\"A Sarajlija, L Armengol, A Maver, I Kitic, D Prokic, M Cehic, M S Djuricic, B Peterlin\",\"doi\":\"10.2478/bjmg-2022-0010\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Deficiency of lysosomal acid lipase (LAL-D) is caused by biallelic pathogenic variants in the <i>LIPA</i> gene. Spectrum of LAL-D ranges from early onset of hepatosplenomegaly and psychomotor regression (Wolman disease) to a more chronic course (cholesteryl ester storage disease - CESD). The diagnosis is based on lipid and biomarker profiles, specific liver histopathology, enzyme deficiency, and identification of causative genetic variants. Biomarker findings are a useful for diagnostics of LAL-D, including high plasma concentration of chitotriosidase as well as elevated oxysterols. Current treatment options include enzyme replacement therapy (sebelipase-alpha), statins, liver transplantation, and stem cell transplantation. We present two pairs of siblings from Serbia with a distinctive phenotype resembling LAL-D with a novel variant of unknown significance (VUS) detected in the <i>LIPA</i> gene and residual LAL activity. All patients presented with hepatosplenomegaly at early childhood. In siblings from family 1, compound heterozygosity for a pathogenic c.419G>A (p.Trp140Ter) variant and a novel VUS c.851C>T (p.Ser284Phe) was detected. Patients from family 2 were homozygous for c.851C>T VUS and both have typical histopathologic findings for LAL-D in the liver. Enzyme activity of LAL was tested in three patients and reported as sufficient, and therefore enzyme replacement therapy could not be approved. When confronted with a challenge of diagnosing an inherited metabolic disorder, several aspects are taken into consideration: clinical manifestations, specific biomarkers, enzyme assay results, and molecular genetic findings. This report brings cases to light which have a considerable discrepancy between those aspects, namely the preserved LAL enzyme activity in presence of clinical manifestations and rare variants in the <i>LIPA</i> gene.</p>\",\"PeriodicalId\":55403,\"journal\":{\"name\":\"Balkan Journal of Medical Genetics\",\"volume\":\"25 1\",\"pages\":\"93-100\"},\"PeriodicalIF\":0.9000,\"publicationDate\":\"2022-06-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/0d/f0/bjmg-25-093.PMC9985358.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Balkan Journal of Medical Genetics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.2478/bjmg-2022-0010\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Balkan Journal of Medical Genetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.2478/bjmg-2022-0010","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
摘要
溶酶体酸性脂肪酶(LAL-D)的缺乏是由LIPA基因的双等位致病变异引起的。LAL-D的频谱范围从早期发病的肝脾肿大和精神运动减退(沃尔曼病)到更慢性的病程(胆固醇酯沉积病- CESD)。诊断是基于脂质和生物标志物概况,特定的肝脏组织病理学,酶缺乏,并确定致病的遗传变异。生物标志物的发现对LAL-D的诊断是有用的,包括高血浆壳三醇苷酶浓度和高氧甾醇。目前的治疗方案包括酶替代疗法(脂酶- α)、他汀类药物、肝移植和干细胞移植。我们介绍了来自塞尔维亚的两对兄弟姐妹,他们具有与LAL- d相似的独特表型,在LIPA基因和残余LAL活性中检测到一种未知意义的新变体(VUS)。所有患者均在儿童早期表现为肝脾肿大。在家族1的兄弟姐妹中,检测到致病性c.419G> a (p.Trp140Ter)变异和新的VUS c.851C>T (p.Ser284Phe)的复合杂合性。来自家族2的患者c.851C>T VUS为纯合子,并且在肝脏中都有典型的LAL-D组织病理学表现。在三名患者中检测了LAL的酶活性,并报告为足够的,因此酶替代疗法不能被批准。当面临诊断遗传性代谢紊乱的挑战时,需要考虑几个方面:临床表现、特定生物标志物、酶测定结果和分子遗传学发现。本报告揭示的病例在这些方面存在相当大的差异,即临床表现中保留的LAL酶活性和LIPA基因的罕见变异。
A Novel Variant in the LIPA Gene Associated with Distinct Phenotype.
Deficiency of lysosomal acid lipase (LAL-D) is caused by biallelic pathogenic variants in the LIPA gene. Spectrum of LAL-D ranges from early onset of hepatosplenomegaly and psychomotor regression (Wolman disease) to a more chronic course (cholesteryl ester storage disease - CESD). The diagnosis is based on lipid and biomarker profiles, specific liver histopathology, enzyme deficiency, and identification of causative genetic variants. Biomarker findings are a useful for diagnostics of LAL-D, including high plasma concentration of chitotriosidase as well as elevated oxysterols. Current treatment options include enzyme replacement therapy (sebelipase-alpha), statins, liver transplantation, and stem cell transplantation. We present two pairs of siblings from Serbia with a distinctive phenotype resembling LAL-D with a novel variant of unknown significance (VUS) detected in the LIPA gene and residual LAL activity. All patients presented with hepatosplenomegaly at early childhood. In siblings from family 1, compound heterozygosity for a pathogenic c.419G>A (p.Trp140Ter) variant and a novel VUS c.851C>T (p.Ser284Phe) was detected. Patients from family 2 were homozygous for c.851C>T VUS and both have typical histopathologic findings for LAL-D in the liver. Enzyme activity of LAL was tested in three patients and reported as sufficient, and therefore enzyme replacement therapy could not be approved. When confronted with a challenge of diagnosing an inherited metabolic disorder, several aspects are taken into consideration: clinical manifestations, specific biomarkers, enzyme assay results, and molecular genetic findings. This report brings cases to light which have a considerable discrepancy between those aspects, namely the preserved LAL enzyme activity in presence of clinical manifestations and rare variants in the LIPA gene.
期刊介绍:
Balkan Journal of Medical Genetics is a journal in the English language for publication of articles involving all branches of medical genetics: human cytogenetics, molecular genetics, clinical genetics, immunogenetics, oncogenetics, pharmacogenetics, population genetics, genetic screening and diagnosis of monogenic and polygenic diseases, prenatal and preimplantation genetic diagnosis, genetic counselling, advances in treatment and prevention.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


