Chenlu Ke, Dipankar Bandyopadhyay, Mario Acunzo, Robert Winn
{"title":"高通量右删减肺癌数据的基因筛选。","authors":"Chenlu Ke, Dipankar Bandyopadhyay, Mario Acunzo, Robert Winn","doi":"10.3390/onco2040017","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Advances in sequencing technologies have allowed collection of massive genome-wide information that substantially advances lung cancer diagnosis and prognosis. Identifying influential markers for clinical endpoints of interest has been an indispensable and critical component of the statistical analysis pipeline. However, classical variable selection methods are not feasible or reliable for high-throughput genetic data. Our objective is to propose a model-free gene screening procedure for high-throughput right-censored data, and to develop a predictive gene signature for lung squamous cell carcinoma (LUSC) with the proposed procedure.</p><p><strong>Methods: </strong>A gene screening procedure was developed based on a recently proposed independence measure. The Cancer Genome Atlas (TCGA) data on LUSC was then studied. The screening procedure was conducted to narrow down the set of influential genes to 378 candidates. A penalized Cox model was then fitted to the reduced set, which further identified a 6-gene signature for LUSC prognosis. The 6-gene signature was validated on datasets from the Gene Expression Omnibus.</p><p><strong>Results: </strong>Both model-fitting and validation results reveal that our method selected influential genes that lead to biologically sensible findings as well as better predictive performance, compared to existing alternatives. According to our multivariable Cox regression analysis, the 6-gene signature was indeed a significant prognostic factor (<i>p</i>-value < 0.001) while controlling for clinical covariates.</p><p><strong>Conclusions: </strong>Gene screening as a fast dimension reduction technique plays an important role in analyzing high-throughput data. The main contribution of this paper is to introduce a fundamental yet pragmatic model-free gene screening approach that aids statistical analysis of right-censored cancer data, and provide a lateral comparison with other available methods in the context of LUSC.</p>","PeriodicalId":74339,"journal":{"name":"Onco","volume":"2 4","pages":"305-318"},"PeriodicalIF":0.0000,"publicationDate":"2022-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10100230/pdf/","citationCount":"0","resultStr":"{\"title\":\"Gene Screening in High-Throughput Right-Censored Lung Cancer Data.\",\"authors\":\"Chenlu Ke, Dipankar Bandyopadhyay, Mario Acunzo, Robert Winn\",\"doi\":\"10.3390/onco2040017\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Advances in sequencing technologies have allowed collection of massive genome-wide information that substantially advances lung cancer diagnosis and prognosis. Identifying influential markers for clinical endpoints of interest has been an indispensable and critical component of the statistical analysis pipeline. However, classical variable selection methods are not feasible or reliable for high-throughput genetic data. Our objective is to propose a model-free gene screening procedure for high-throughput right-censored data, and to develop a predictive gene signature for lung squamous cell carcinoma (LUSC) with the proposed procedure.</p><p><strong>Methods: </strong>A gene screening procedure was developed based on a recently proposed independence measure. The Cancer Genome Atlas (TCGA) data on LUSC was then studied. The screening procedure was conducted to narrow down the set of influential genes to 378 candidates. A penalized Cox model was then fitted to the reduced set, which further identified a 6-gene signature for LUSC prognosis. The 6-gene signature was validated on datasets from the Gene Expression Omnibus.</p><p><strong>Results: </strong>Both model-fitting and validation results reveal that our method selected influential genes that lead to biologically sensible findings as well as better predictive performance, compared to existing alternatives. According to our multivariable Cox regression analysis, the 6-gene signature was indeed a significant prognostic factor (<i>p</i>-value < 0.001) while controlling for clinical covariates.</p><p><strong>Conclusions: </strong>Gene screening as a fast dimension reduction technique plays an important role in analyzing high-throughput data. The main contribution of this paper is to introduce a fundamental yet pragmatic model-free gene screening approach that aids statistical analysis of right-censored cancer data, and provide a lateral comparison with other available methods in the context of LUSC.</p>\",\"PeriodicalId\":74339,\"journal\":{\"name\":\"Onco\",\"volume\":\"2 4\",\"pages\":\"305-318\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2022-12-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10100230/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Onco\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.3390/onco2040017\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Onco","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3390/onco2040017","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
Gene Screening in High-Throughput Right-Censored Lung Cancer Data.
Background: Advances in sequencing technologies have allowed collection of massive genome-wide information that substantially advances lung cancer diagnosis and prognosis. Identifying influential markers for clinical endpoints of interest has been an indispensable and critical component of the statistical analysis pipeline. However, classical variable selection methods are not feasible or reliable for high-throughput genetic data. Our objective is to propose a model-free gene screening procedure for high-throughput right-censored data, and to develop a predictive gene signature for lung squamous cell carcinoma (LUSC) with the proposed procedure.
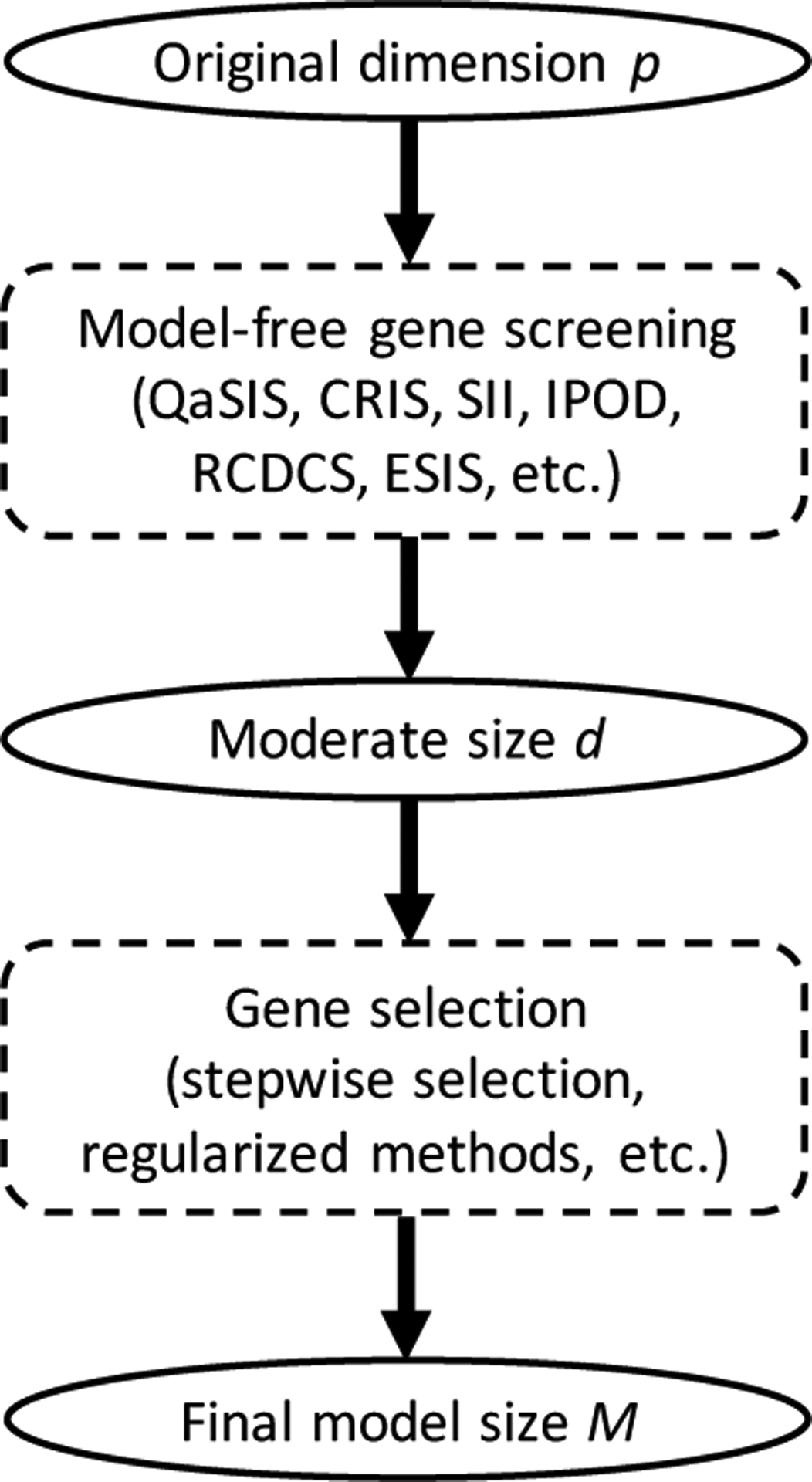
Methods: A gene screening procedure was developed based on a recently proposed independence measure. The Cancer Genome Atlas (TCGA) data on LUSC was then studied. The screening procedure was conducted to narrow down the set of influential genes to 378 candidates. A penalized Cox model was then fitted to the reduced set, which further identified a 6-gene signature for LUSC prognosis. The 6-gene signature was validated on datasets from the Gene Expression Omnibus.
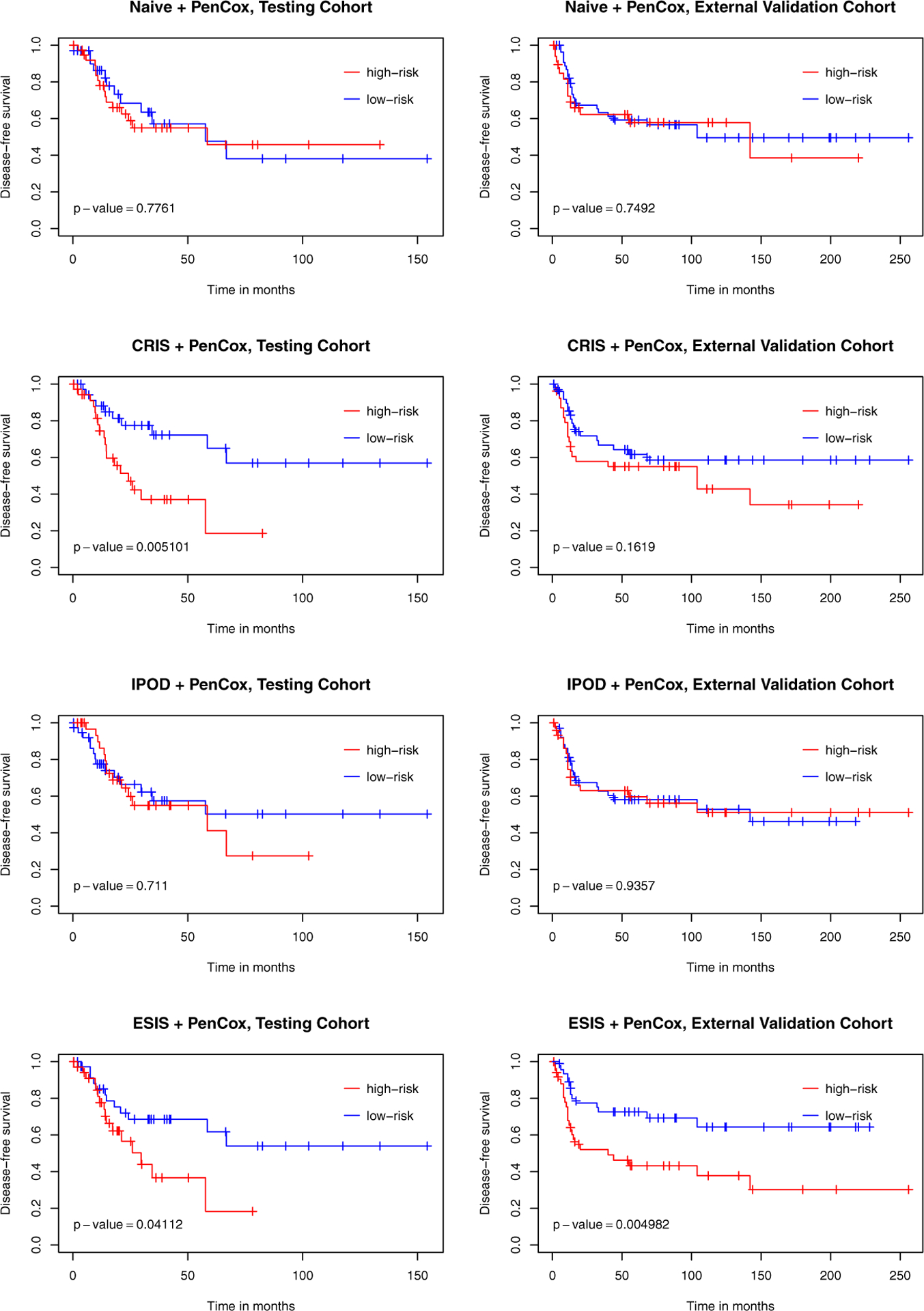
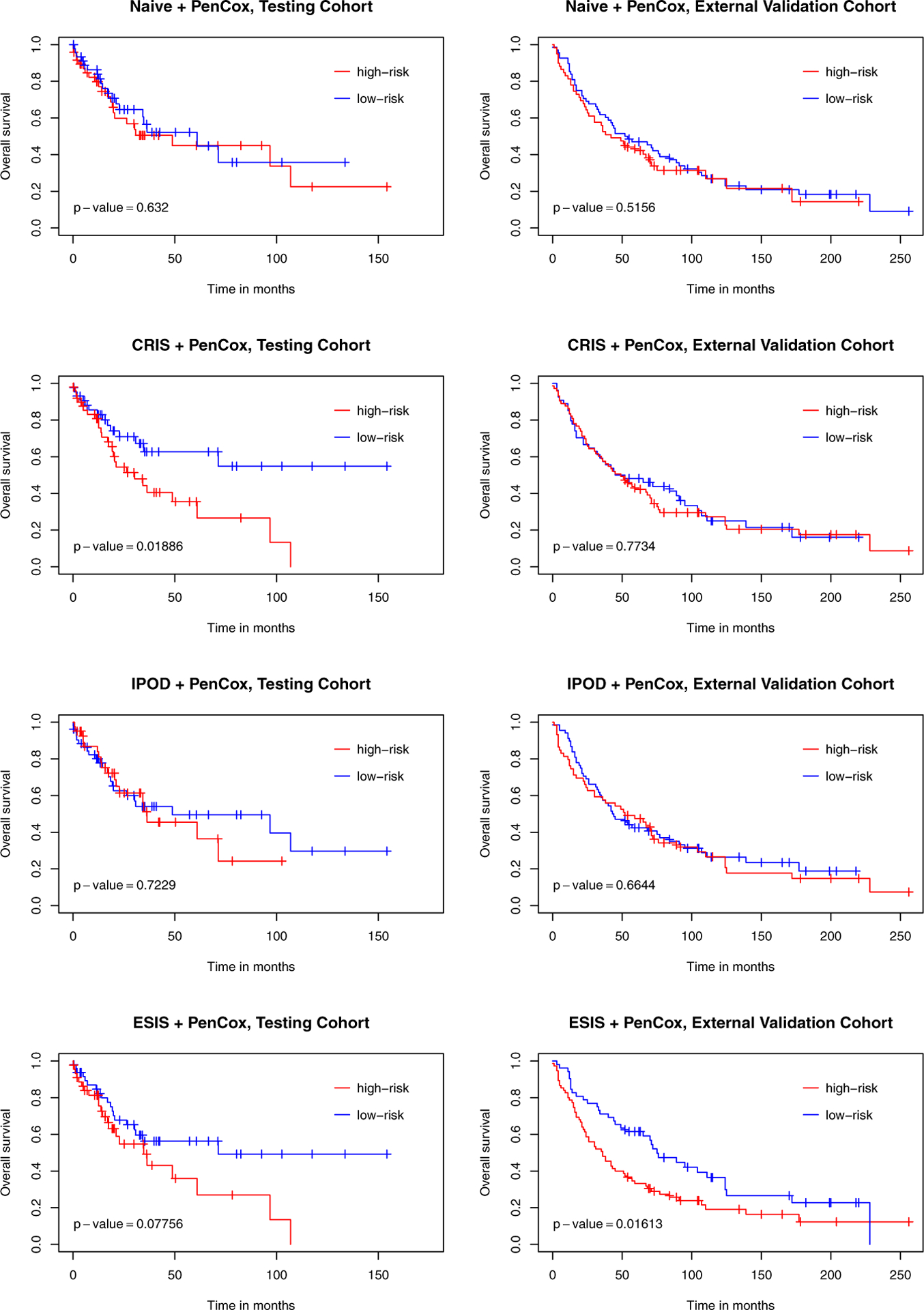
Results: Both model-fitting and validation results reveal that our method selected influential genes that lead to biologically sensible findings as well as better predictive performance, compared to existing alternatives. According to our multivariable Cox regression analysis, the 6-gene signature was indeed a significant prognostic factor (p-value < 0.001) while controlling for clinical covariates.
Conclusions: Gene screening as a fast dimension reduction technique plays an important role in analyzing high-throughput data. The main contribution of this paper is to introduce a fundamental yet pragmatic model-free gene screening approach that aids statistical analysis of right-censored cancer data, and provide a lateral comparison with other available methods in the context of LUSC.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


