M Y Lobanov, M V Slizen, N V Dovidchenko, A V Panfilov, A A Surin, I V Likhachev, O V Galzitskaya
{"title":"深度学习模型与简单方法的比较,以评估抗菌肽预测问题。","authors":"M Y Lobanov, M V Slizen, N V Dovidchenko, A V Panfilov, A A Surin, I V Likhachev, O V Galzitskaya","doi":"10.1002/minf.202200181","DOIUrl":null,"url":null,"abstract":"<p><p>Antibiotic-resistant strains are an emerging threat to public health. The usage of antimicrobial peptides (AMPs) is one of the promising approaches to solve this problem. For the development of new AMPs, it is necessary to have reliable prediction methods. Recently, deep learning approaches have been used to predict AMP. In this paper, we want to compare simple and complex methods for these purposes. We used the BERT transformer to create sequence embeddings and the multilayer perceptron (MLP) and light attention (LA) approaches for classification. One of them reached about 80 % accuracy and specificity in benchmark testing, which is on par with the best available methods. For comparison, we proposed a simple method using only the amino acid composition of proteins or peptides. This method has shown good results, at the level of the best methods. We have prepared a special server for predicting the ability of AMPs by amino acid composition: http://bioproteom.protres.ru/antimicrob/.</p>","PeriodicalId":18853,"journal":{"name":"Molecular Informatics","volume":" ","pages":"e202200181"},"PeriodicalIF":3.1000,"publicationDate":"2024-05-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Comparison of deep learning models with simple method to assess the problem of antimicrobial peptides prediction.\",\"authors\":\"M Y Lobanov, M V Slizen, N V Dovidchenko, A V Panfilov, A A Surin, I V Likhachev, O V Galzitskaya\",\"doi\":\"10.1002/minf.202200181\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Antibiotic-resistant strains are an emerging threat to public health. The usage of antimicrobial peptides (AMPs) is one of the promising approaches to solve this problem. For the development of new AMPs, it is necessary to have reliable prediction methods. Recently, deep learning approaches have been used to predict AMP. In this paper, we want to compare simple and complex methods for these purposes. We used the BERT transformer to create sequence embeddings and the multilayer perceptron (MLP) and light attention (LA) approaches for classification. One of them reached about 80 % accuracy and specificity in benchmark testing, which is on par with the best available methods. For comparison, we proposed a simple method using only the amino acid composition of proteins or peptides. This method has shown good results, at the level of the best methods. We have prepared a special server for predicting the ability of AMPs by amino acid composition: http://bioproteom.protres.ru/antimicrob/.</p>\",\"PeriodicalId\":18853,\"journal\":{\"name\":\"Molecular Informatics\",\"volume\":\" \",\"pages\":\"e202200181\"},\"PeriodicalIF\":3.1000,\"publicationDate\":\"2024-05-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Molecular Informatics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1002/minf.202200181\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/4/7 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, MEDICINAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Informatics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1002/minf.202200181","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/4/7 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
Comparison of deep learning models with simple method to assess the problem of antimicrobial peptides prediction.
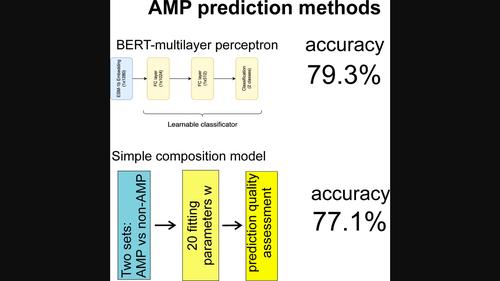
Antibiotic-resistant strains are an emerging threat to public health. The usage of antimicrobial peptides (AMPs) is one of the promising approaches to solve this problem. For the development of new AMPs, it is necessary to have reliable prediction methods. Recently, deep learning approaches have been used to predict AMP. In this paper, we want to compare simple and complex methods for these purposes. We used the BERT transformer to create sequence embeddings and the multilayer perceptron (MLP) and light attention (LA) approaches for classification. One of them reached about 80 % accuracy and specificity in benchmark testing, which is on par with the best available methods. For comparison, we proposed a simple method using only the amino acid composition of proteins or peptides. This method has shown good results, at the level of the best methods. We have prepared a special server for predicting the ability of AMPs by amino acid composition: http://bioproteom.protres.ru/antimicrob/.
期刊介绍:
Molecular Informatics is a peer-reviewed, international forum for publication of high-quality, interdisciplinary research on all molecular aspects of bio/cheminformatics and computer-assisted molecular design. Molecular Informatics succeeded QSAR & Combinatorial Science in 2010.
Molecular Informatics presents methodological innovations that will lead to a deeper understanding of ligand-receptor interactions, macromolecular complexes, molecular networks, design concepts and processes that demonstrate how ideas and design concepts lead to molecules with a desired structure or function, preferably including experimental validation.
The journal''s scope includes but is not limited to the fields of drug discovery and chemical biology, protein and nucleic acid engineering and design, the design of nanomolecular structures, strategies for modeling of macromolecular assemblies, molecular networks and systems, pharmaco- and chemogenomics, computer-assisted screening strategies, as well as novel technologies for the de novo design of biologically active molecules. As a unique feature Molecular Informatics publishes so-called "Methods Corner" review-type articles which feature important technological concepts and advances within the scope of the journal.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


