{"title":"比较基因组学和信息含量分析揭示了假单胞菌属中具有基因组水平分辨率的MLSA的核心基因rpoD、pepN和gltX的内部区域","authors":"Matías Garavaglia , Andrés Muzlera , Claudio Valverde","doi":"10.1016/j.ympev.2022.107663","DOIUrl":null,"url":null,"abstract":"<div><p>In the field of prokaryotic taxonomy, there has been a recent transition towards phylogenomics as the gold standard approach. However, genome-based phylogenetics is still restrictive for its cost when managing large amounts of isolates. Fast, cheap, and taxonomically competent alternatives, like multilocus sequence analysis (MLSA) are thus recommendable. Nevertheless, the criteria for selecting the conserved genes for MLSA have not been explicit for different bacterial taxa, including the broadly diverse <em>Pseudomonas</em> genus. Here, we have carried out an unbiased and rational workflow to select internal sequence regions of <em>Pseudomonas</em> core genes (CG) for a MLSA with the best phylogenetic power, and with a resolution comparable to the genome-based ANI approach. A computational workflow was established to inspect 126 complete genomes of representatives from over 60 <em>Pseudomonas</em> species and subspecies, in order to identify the most informative CG internal regions and determine which combinations in sets of three partial CG sequences have comparable phylogenetic resolution to that of the current ANI standard. We found that the <em>rpoD</em><sup>346-1196</sup>-<em>pepN</em><sup>1711-2571</sup>-<em>gltX</em><sup>86-909</sup> concatenated sequences were the best performing in terms of phylogenetic robustness and resulted highly sensitive and specific when contrasted with ANI. The <em>rpoD</em>-<em>pepN</em>-<em>gltX</em> MLSA was validated <em>in silico</em> and <em>in vitro</em>. Altogether, the results presented here supports the proposal of the <em>rpoD</em>-<em>pepN</em>-<em>gltX</em> MLSA as a fast, affordable, and robust phylogenetic tool for members of the <em>Pseudomonas</em> genus.</p></div>","PeriodicalId":56109,"journal":{"name":"Molecular Phylogenetics and Evolution","volume":"179 ","pages":"Article 107663"},"PeriodicalIF":3.6000,"publicationDate":"2023-02-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"1","resultStr":"{\"title\":\"Comparative genomics and informational content analysis uncovered internal regions of the core genes rpoD, pepN and gltX for an MLSA with genome-level resolving power within the genus Pseudomonas\",\"authors\":\"Matías Garavaglia , Andrés Muzlera , Claudio Valverde\",\"doi\":\"10.1016/j.ympev.2022.107663\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>In the field of prokaryotic taxonomy, there has been a recent transition towards phylogenomics as the gold standard approach. However, genome-based phylogenetics is still restrictive for its cost when managing large amounts of isolates. Fast, cheap, and taxonomically competent alternatives, like multilocus sequence analysis (MLSA) are thus recommendable. Nevertheless, the criteria for selecting the conserved genes for MLSA have not been explicit for different bacterial taxa, including the broadly diverse <em>Pseudomonas</em> genus. Here, we have carried out an unbiased and rational workflow to select internal sequence regions of <em>Pseudomonas</em> core genes (CG) for a MLSA with the best phylogenetic power, and with a resolution comparable to the genome-based ANI approach. A computational workflow was established to inspect 126 complete genomes of representatives from over 60 <em>Pseudomonas</em> species and subspecies, in order to identify the most informative CG internal regions and determine which combinations in sets of three partial CG sequences have comparable phylogenetic resolution to that of the current ANI standard. We found that the <em>rpoD</em><sup>346-1196</sup>-<em>pepN</em><sup>1711-2571</sup>-<em>gltX</em><sup>86-909</sup> concatenated sequences were the best performing in terms of phylogenetic robustness and resulted highly sensitive and specific when contrasted with ANI. The <em>rpoD</em>-<em>pepN</em>-<em>gltX</em> MLSA was validated <em>in silico</em> and <em>in vitro</em>. Altogether, the results presented here supports the proposal of the <em>rpoD</em>-<em>pepN</em>-<em>gltX</em> MLSA as a fast, affordable, and robust phylogenetic tool for members of the <em>Pseudomonas</em> genus.</p></div>\",\"PeriodicalId\":56109,\"journal\":{\"name\":\"Molecular Phylogenetics and Evolution\",\"volume\":\"179 \",\"pages\":\"Article 107663\"},\"PeriodicalIF\":3.6000,\"publicationDate\":\"2023-02-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"1\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Molecular Phylogenetics and Evolution\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S1055790322002767\",\"RegionNum\":1,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Phylogenetics and Evolution","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S1055790322002767","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
Comparative genomics and informational content analysis uncovered internal regions of the core genes rpoD, pepN and gltX for an MLSA with genome-level resolving power within the genus Pseudomonas
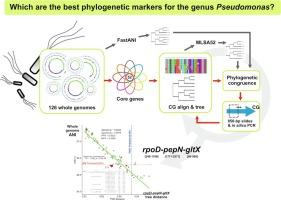
In the field of prokaryotic taxonomy, there has been a recent transition towards phylogenomics as the gold standard approach. However, genome-based phylogenetics is still restrictive for its cost when managing large amounts of isolates. Fast, cheap, and taxonomically competent alternatives, like multilocus sequence analysis (MLSA) are thus recommendable. Nevertheless, the criteria for selecting the conserved genes for MLSA have not been explicit for different bacterial taxa, including the broadly diverse Pseudomonas genus. Here, we have carried out an unbiased and rational workflow to select internal sequence regions of Pseudomonas core genes (CG) for a MLSA with the best phylogenetic power, and with a resolution comparable to the genome-based ANI approach. A computational workflow was established to inspect 126 complete genomes of representatives from over 60 Pseudomonas species and subspecies, in order to identify the most informative CG internal regions and determine which combinations in sets of three partial CG sequences have comparable phylogenetic resolution to that of the current ANI standard. We found that the rpoD346-1196-pepN1711-2571-gltX86-909 concatenated sequences were the best performing in terms of phylogenetic robustness and resulted highly sensitive and specific when contrasted with ANI. The rpoD-pepN-gltX MLSA was validated in silico and in vitro. Altogether, the results presented here supports the proposal of the rpoD-pepN-gltX MLSA as a fast, affordable, and robust phylogenetic tool for members of the Pseudomonas genus.
期刊介绍:
Molecular Phylogenetics and Evolution is dedicated to bringing Darwin''s dream within grasp - to "have fairly true genealogical trees of each great kingdom of Nature." The journal provides a forum for molecular studies that advance our understanding of phylogeny and evolution, further the development of phylogenetically more accurate taxonomic classifications, and ultimately bring a unified classification for all the ramifying lines of life. Phylogeographic studies will be considered for publication if they offer EXCEPTIONAL theoretical or empirical advances.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


