René N Caballero-Florán, Kendall P Dean, Andrew D Nelson, Lia Min, Paul M Jenkins
{"title":"慢性锂治疗对Ank3突变小鼠模型神经元兴奋性和gaba能传递的影响。","authors":"René N Caballero-Florán, Kendall P Dean, Andrew D Nelson, Lia Min, Paul M Jenkins","doi":"10.1101/2023.10.26.564203","DOIUrl":null,"url":null,"abstract":"<p><p>Bipolar disorder (BD) is a prevalent psychiatric condition characterized by mood dysregulation, psychosocial impairment, and an increased risk of suicide. The gene <i>ANK3</i> has been identified as a risk locus for BD through multiple genome-wide association studies (GWAS). However, the mechanisms by which <i>ANK3</i> variants influence BD pathophysiology and treatment response remain unclear. <i>ANK3</i> encodes ankyrin-G, a protein that organizes the axon initial segment (AIS) and nodes of Ranvier by scaffolding ion channels and cell adhesion molecules to the cytoskeleton. Recent studies show that ankyrin-G interacts with the GABA<sub>A</sub> receptor-associated protein (GABARAP) to stabilize inhibitory synapses, potentially linking <i>ANK3</i> variants to inhibitory (GABAergic) signaling deficits associated with BD. We previously demonstrated that the BD-associated variant, <i>ANK3</i> p.W1989R, disrupts the ankyrin-G/GABARAP interaction, resulting in inhibitory deficits and cortical pyramidal neuron hyperexcitability in mice. In this study, we investigate how lithium, a common BD therapeutic, modulates neuronal excitability in this model. Our findings show that chronic lithium treatment selectively enhances presynaptic GABAergic neurotransmission, reduces neuronal hyperexcitability, and partially rescues AIS length, without altering the density of GABAergic synapses. We also show that the selective glycogen synthase kinase-3 beta (GSK-3β) inhibitor Tideglusib recapitulates the enhancement of presynaptic GABAergic signaling. These findings shed new light on how <i>ANK3</i> variants may contribute to inhibitory deficits in BD and demonstrate that lithium treatment is able to restore these deficits, likely through GSK-3β inhibition. Furthermore, these findings highlight GSK-3β inhibition as a promising therapeutic strategy for treating BD and other neurological disorders affected by GABAergic dysfunction.</p>","PeriodicalId":72407,"journal":{"name":"bioRxiv : the preprint server for biology","volume":" ","pages":""},"PeriodicalIF":0.0000,"publicationDate":"2025-07-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10634991/pdf/","citationCount":"0","resultStr":"{\"title\":\"Lithium Restores Inhibitory Function and Neuronal Excitability through GSK-3β Inhibition in a Bipolar Disorder-Associated <i>Ank3</i> Variant Mouse Model.\",\"authors\":\"René N Caballero-Florán, Kendall P Dean, Andrew D Nelson, Lia Min, Paul M Jenkins\",\"doi\":\"10.1101/2023.10.26.564203\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Bipolar disorder (BD) is a prevalent psychiatric condition characterized by mood dysregulation, psychosocial impairment, and an increased risk of suicide. The gene <i>ANK3</i> has been identified as a risk locus for BD through multiple genome-wide association studies (GWAS). However, the mechanisms by which <i>ANK3</i> variants influence BD pathophysiology and treatment response remain unclear. <i>ANK3</i> encodes ankyrin-G, a protein that organizes the axon initial segment (AIS) and nodes of Ranvier by scaffolding ion channels and cell adhesion molecules to the cytoskeleton. Recent studies show that ankyrin-G interacts with the GABA<sub>A</sub> receptor-associated protein (GABARAP) to stabilize inhibitory synapses, potentially linking <i>ANK3</i> variants to inhibitory (GABAergic) signaling deficits associated with BD. We previously demonstrated that the BD-associated variant, <i>ANK3</i> p.W1989R, disrupts the ankyrin-G/GABARAP interaction, resulting in inhibitory deficits and cortical pyramidal neuron hyperexcitability in mice. In this study, we investigate how lithium, a common BD therapeutic, modulates neuronal excitability in this model. Our findings show that chronic lithium treatment selectively enhances presynaptic GABAergic neurotransmission, reduces neuronal hyperexcitability, and partially rescues AIS length, without altering the density of GABAergic synapses. We also show that the selective glycogen synthase kinase-3 beta (GSK-3β) inhibitor Tideglusib recapitulates the enhancement of presynaptic GABAergic signaling. These findings shed new light on how <i>ANK3</i> variants may contribute to inhibitory deficits in BD and demonstrate that lithium treatment is able to restore these deficits, likely through GSK-3β inhibition. Furthermore, these findings highlight GSK-3β inhibition as a promising therapeutic strategy for treating BD and other neurological disorders affected by GABAergic dysfunction.</p>\",\"PeriodicalId\":72407,\"journal\":{\"name\":\"bioRxiv : the preprint server for biology\",\"volume\":\" \",\"pages\":\"\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2025-07-02\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10634991/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"bioRxiv : the preprint server for biology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1101/2023.10.26.564203\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"bioRxiv : the preprint server for biology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1101/2023.10.26.564203","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
Lithium Restores Inhibitory Function and Neuronal Excitability through GSK-3β Inhibition in a Bipolar Disorder-Associated Ank3 Variant Mouse Model.
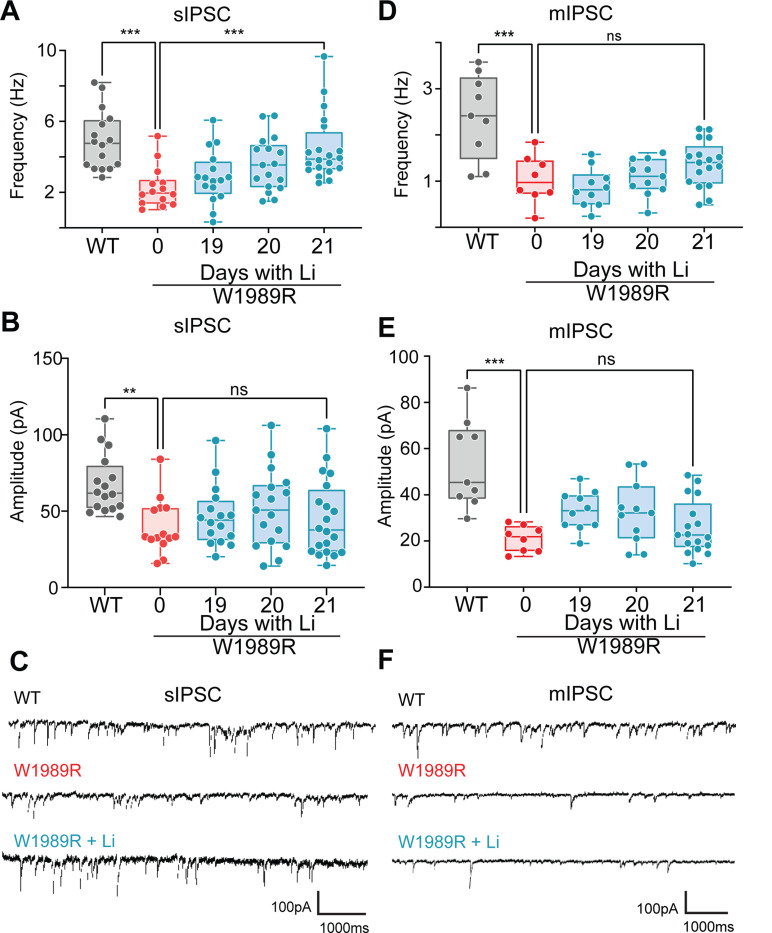
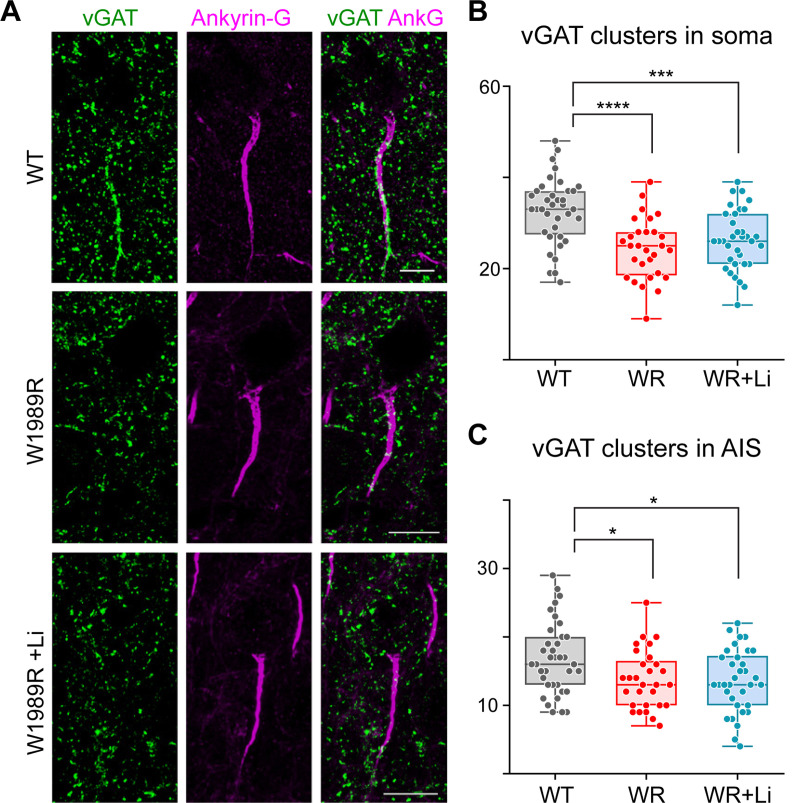
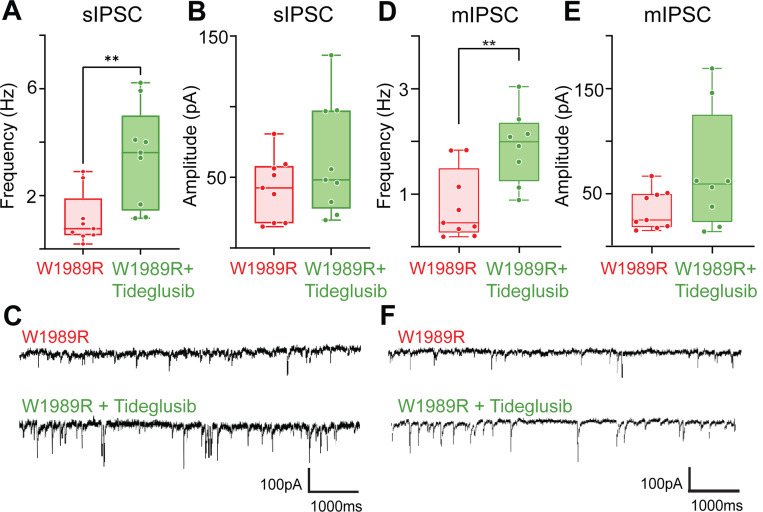
Bipolar disorder (BD) is a prevalent psychiatric condition characterized by mood dysregulation, psychosocial impairment, and an increased risk of suicide. The gene ANK3 has been identified as a risk locus for BD through multiple genome-wide association studies (GWAS). However, the mechanisms by which ANK3 variants influence BD pathophysiology and treatment response remain unclear. ANK3 encodes ankyrin-G, a protein that organizes the axon initial segment (AIS) and nodes of Ranvier by scaffolding ion channels and cell adhesion molecules to the cytoskeleton. Recent studies show that ankyrin-G interacts with the GABAA receptor-associated protein (GABARAP) to stabilize inhibitory synapses, potentially linking ANK3 variants to inhibitory (GABAergic) signaling deficits associated with BD. We previously demonstrated that the BD-associated variant, ANK3 p.W1989R, disrupts the ankyrin-G/GABARAP interaction, resulting in inhibitory deficits and cortical pyramidal neuron hyperexcitability in mice. In this study, we investigate how lithium, a common BD therapeutic, modulates neuronal excitability in this model. Our findings show that chronic lithium treatment selectively enhances presynaptic GABAergic neurotransmission, reduces neuronal hyperexcitability, and partially rescues AIS length, without altering the density of GABAergic synapses. We also show that the selective glycogen synthase kinase-3 beta (GSK-3β) inhibitor Tideglusib recapitulates the enhancement of presynaptic GABAergic signaling. These findings shed new light on how ANK3 variants may contribute to inhibitory deficits in BD and demonstrate that lithium treatment is able to restore these deficits, likely through GSK-3β inhibition. Furthermore, these findings highlight GSK-3β inhibition as a promising therapeutic strategy for treating BD and other neurological disorders affected by GABAergic dysfunction.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


