Nourelislam Awad, Rania Hassan Mohamed, Nehal I Ghoneim, Ahmed O Elmehrath, Nagwa El-Badri
{"title":"SARS-CoV-2抗原表位预测的免疫信息学方法。","authors":"Nourelislam Awad, Rania Hassan Mohamed, Nehal I Ghoneim, Ahmed O Elmehrath, Nagwa El-Badri","doi":"10.1186/s43141-022-00344-1","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>The novel coronavirus (SARS-CoV-2) caused lethal infections worldwide during an unprecedented pandemic. Identification of the candidate viral epitopes is the first step in the design of vaccines against the viral infection. Several immunoinformatic approaches were employed to identify the SARS-CoV-2 epitopes that bind specifically with the major histocompatibility molecules class I (MHC-I). We utilized immunoinformatic tools to analyze the whole viral protein sequences, to identify the SARS-CoV-2 epitopes responsible for binding to the most frequent human leukocyte antigen (HLA) alleles in the Egyptian population. These alleles were also found with high frequency in other populations worldwide.</p><p><strong>Results: </strong>Molecular docking approach showed that using the co-crystallized MHC-I and T cell receptor (TCR) instead of using MHC-I structure only, significantly enhanced docking scores and stabilized the conformation, as well as the binding affinity of the identified SARS-CoV-2 epitopes. Our approach directly predicts 7 potential vaccine subunits from the available SARS-CoV-2 spike and ORF1ab protein sequence. This prediction has been confirmed by published experimentally validated and in silico predicted spike epitope. On the other hand, we predicted novel epitopes (RDLPQGFSA and FCLEASFNY) showing high docking scores and antigenicity response with both MHC-I and TCR. Moreover, antigenicity, allergenicity, toxicity, and physicochemical properties of the predicted SARS-CoV-2 epitopes were evaluated via state-of-the-art bioinformatic approaches, showing high efficacy of the proposed epitopes as a vaccine candidate.</p><p><strong>Conclusion: </strong>Our predicted SARS-CoV-2 epitopes can facilitate vaccine development to enhance the immunogenicity against SARS-CoV-2 and provide supportive data for further experimental validation. Our proposed molecular docking approach of exploiting both MHC and TCR structures can be used to identify potential epitopes for most microbial pathogens, provided the crystal structure of MHC co-crystallized with TCR.</p>","PeriodicalId":74026,"journal":{"name":"Journal, genetic engineering & biotechnology","volume":"20 1","pages":"60"},"PeriodicalIF":2.8000,"publicationDate":"2022-04-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9019534/pdf/","citationCount":"3","resultStr":"{\"title\":\"Immunoinformatics approach of epitope prediction for SARS-CoV-2.\",\"authors\":\"Nourelislam Awad, Rania Hassan Mohamed, Nehal I Ghoneim, Ahmed O Elmehrath, Nagwa El-Badri\",\"doi\":\"10.1186/s43141-022-00344-1\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>The novel coronavirus (SARS-CoV-2) caused lethal infections worldwide during an unprecedented pandemic. Identification of the candidate viral epitopes is the first step in the design of vaccines against the viral infection. Several immunoinformatic approaches were employed to identify the SARS-CoV-2 epitopes that bind specifically with the major histocompatibility molecules class I (MHC-I). We utilized immunoinformatic tools to analyze the whole viral protein sequences, to identify the SARS-CoV-2 epitopes responsible for binding to the most frequent human leukocyte antigen (HLA) alleles in the Egyptian population. These alleles were also found with high frequency in other populations worldwide.</p><p><strong>Results: </strong>Molecular docking approach showed that using the co-crystallized MHC-I and T cell receptor (TCR) instead of using MHC-I structure only, significantly enhanced docking scores and stabilized the conformation, as well as the binding affinity of the identified SARS-CoV-2 epitopes. Our approach directly predicts 7 potential vaccine subunits from the available SARS-CoV-2 spike and ORF1ab protein sequence. This prediction has been confirmed by published experimentally validated and in silico predicted spike epitope. On the other hand, we predicted novel epitopes (RDLPQGFSA and FCLEASFNY) showing high docking scores and antigenicity response with both MHC-I and TCR. Moreover, antigenicity, allergenicity, toxicity, and physicochemical properties of the predicted SARS-CoV-2 epitopes were evaluated via state-of-the-art bioinformatic approaches, showing high efficacy of the proposed epitopes as a vaccine candidate.</p><p><strong>Conclusion: </strong>Our predicted SARS-CoV-2 epitopes can facilitate vaccine development to enhance the immunogenicity against SARS-CoV-2 and provide supportive data for further experimental validation. Our proposed molecular docking approach of exploiting both MHC and TCR structures can be used to identify potential epitopes for most microbial pathogens, provided the crystal structure of MHC co-crystallized with TCR.</p>\",\"PeriodicalId\":74026,\"journal\":{\"name\":\"Journal, genetic engineering & biotechnology\",\"volume\":\"20 1\",\"pages\":\"60\"},\"PeriodicalIF\":2.8000,\"publicationDate\":\"2022-04-20\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9019534/pdf/\",\"citationCount\":\"3\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal, genetic engineering & biotechnology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1186/s43141-022-00344-1\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"BIOTECHNOLOGY & APPLIED MICROBIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal, genetic engineering & biotechnology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s43141-022-00344-1","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOTECHNOLOGY & APPLIED MICROBIOLOGY","Score":null,"Total":0}
Immunoinformatics approach of epitope prediction for SARS-CoV-2.
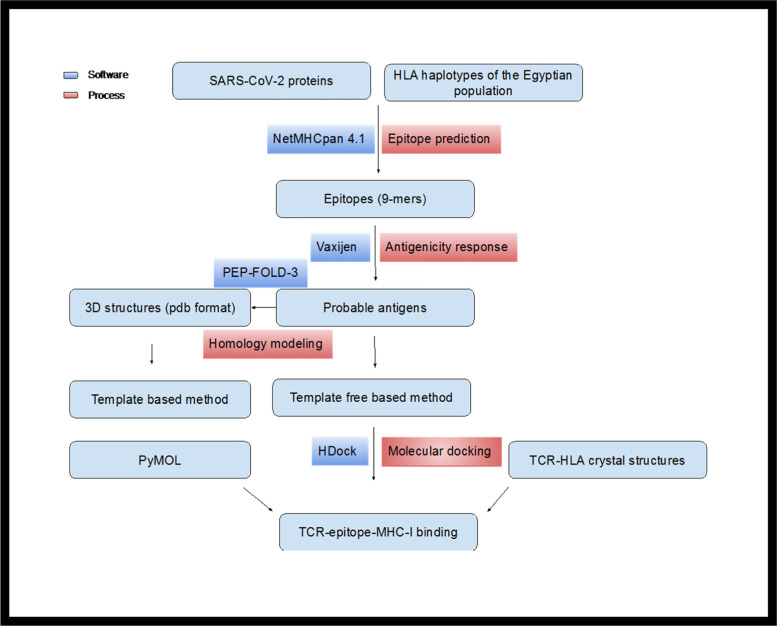
Background: The novel coronavirus (SARS-CoV-2) caused lethal infections worldwide during an unprecedented pandemic. Identification of the candidate viral epitopes is the first step in the design of vaccines against the viral infection. Several immunoinformatic approaches were employed to identify the SARS-CoV-2 epitopes that bind specifically with the major histocompatibility molecules class I (MHC-I). We utilized immunoinformatic tools to analyze the whole viral protein sequences, to identify the SARS-CoV-2 epitopes responsible for binding to the most frequent human leukocyte antigen (HLA) alleles in the Egyptian population. These alleles were also found with high frequency in other populations worldwide.
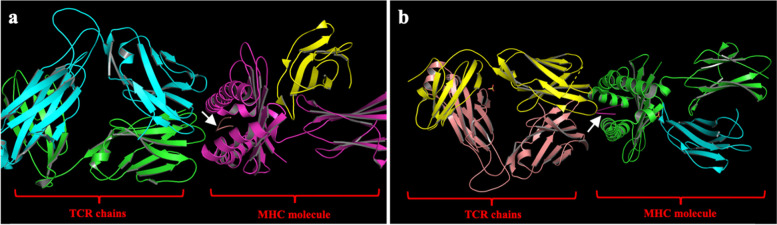
Results: Molecular docking approach showed that using the co-crystallized MHC-I and T cell receptor (TCR) instead of using MHC-I structure only, significantly enhanced docking scores and stabilized the conformation, as well as the binding affinity of the identified SARS-CoV-2 epitopes. Our approach directly predicts 7 potential vaccine subunits from the available SARS-CoV-2 spike and ORF1ab protein sequence. This prediction has been confirmed by published experimentally validated and in silico predicted spike epitope. On the other hand, we predicted novel epitopes (RDLPQGFSA and FCLEASFNY) showing high docking scores and antigenicity response with both MHC-I and TCR. Moreover, antigenicity, allergenicity, toxicity, and physicochemical properties of the predicted SARS-CoV-2 epitopes were evaluated via state-of-the-art bioinformatic approaches, showing high efficacy of the proposed epitopes as a vaccine candidate.
Conclusion: Our predicted SARS-CoV-2 epitopes can facilitate vaccine development to enhance the immunogenicity against SARS-CoV-2 and provide supportive data for further experimental validation. Our proposed molecular docking approach of exploiting both MHC and TCR structures can be used to identify potential epitopes for most microbial pathogens, provided the crystal structure of MHC co-crystallized with TCR.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


