{"title":"空间转录组学的转录组学和组织病理学图像整合分析","authors":"Yiran Shan , Qian Zhang , Wenbo Guo , Yanhong Wu , Yuxin Miao , Hongyi Xin , Qiuyu Lian , Jin Gu","doi":"10.1016/j.gpb.2022.11.012","DOIUrl":null,"url":null,"abstract":"<div><p>Sequencing-based <strong>spatial transcriptomics</strong> (ST) is an emerging technology to study <em>in situ</em> gene expression patterns at the whole-genome scale. Currently, ST data analysis is still complicated by high technical noises and low resolution. In addition to the transcriptomic data, matched histopathological images are usually generated for the same tissue sample along the ST experiment. The matched high-resolution histopathological images provide complementary cellular phenotypical information, providing an opportunity to mitigate the noises in ST data. We present a novel ST data analysis method called transcriptome and histopathological image integrative analysis for ST (TIST), which enables the identification of spatial clusters (SCs) and the enhancement of spatial gene expression patterns by integrative analysis of matched transcriptomic data and images. TIST devises a histopathological feature extraction method based on Markov random field (MRF) to learn the cellular features from histopathological images, and integrates them with the transcriptomic data and location information as a network, termed TIST-net. Based on TIST-net, SCs are identified by a random walk-based strategy, and gene expression patterns are enhanced by neighborhood smoothing. We benchmark TIST on both simulated datasets and 32 real samples against several state-of-the-art methods. Results show that TIST is robust to technical noises on multiple analysis tasks for sequencing-based ST data and can find interesting microstructures in different biological scenarios. TIST is available at <span>http://lifeome.net/software/tist/</span><svg><path></path></svg> and <span>https://ngdc.cncb.ac.cn/biocode/tools/BT007317</span><svg><path></path></svg>.</p></div>","PeriodicalId":12528,"journal":{"name":"Genomics, Proteomics & Bioinformatics","volume":"20 5","pages":"Pages 974-988"},"PeriodicalIF":11.5000,"publicationDate":"2022-10-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10025771/pdf/","citationCount":"0","resultStr":"{\"title\":\"TIST: Transcriptome and Histopathological Image Integrative Analysis for Spatial Transcriptomics\",\"authors\":\"Yiran Shan , Qian Zhang , Wenbo Guo , Yanhong Wu , Yuxin Miao , Hongyi Xin , Qiuyu Lian , Jin Gu\",\"doi\":\"10.1016/j.gpb.2022.11.012\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>Sequencing-based <strong>spatial transcriptomics</strong> (ST) is an emerging technology to study <em>in situ</em> gene expression patterns at the whole-genome scale. Currently, ST data analysis is still complicated by high technical noises and low resolution. In addition to the transcriptomic data, matched histopathological images are usually generated for the same tissue sample along the ST experiment. The matched high-resolution histopathological images provide complementary cellular phenotypical information, providing an opportunity to mitigate the noises in ST data. We present a novel ST data analysis method called transcriptome and histopathological image integrative analysis for ST (TIST), which enables the identification of spatial clusters (SCs) and the enhancement of spatial gene expression patterns by integrative analysis of matched transcriptomic data and images. TIST devises a histopathological feature extraction method based on Markov random field (MRF) to learn the cellular features from histopathological images, and integrates them with the transcriptomic data and location information as a network, termed TIST-net. Based on TIST-net, SCs are identified by a random walk-based strategy, and gene expression patterns are enhanced by neighborhood smoothing. We benchmark TIST on both simulated datasets and 32 real samples against several state-of-the-art methods. Results show that TIST is robust to technical noises on multiple analysis tasks for sequencing-based ST data and can find interesting microstructures in different biological scenarios. TIST is available at <span>http://lifeome.net/software/tist/</span><svg><path></path></svg> and <span>https://ngdc.cncb.ac.cn/biocode/tools/BT007317</span><svg><path></path></svg>.</p></div>\",\"PeriodicalId\":12528,\"journal\":{\"name\":\"Genomics, Proteomics & Bioinformatics\",\"volume\":\"20 5\",\"pages\":\"Pages 974-988\"},\"PeriodicalIF\":11.5000,\"publicationDate\":\"2022-10-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10025771/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Genomics, Proteomics & Bioinformatics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S167202292200167X\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Genomics, Proteomics & Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S167202292200167X","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
TIST: Transcriptome and Histopathological Image Integrative Analysis for Spatial Transcriptomics
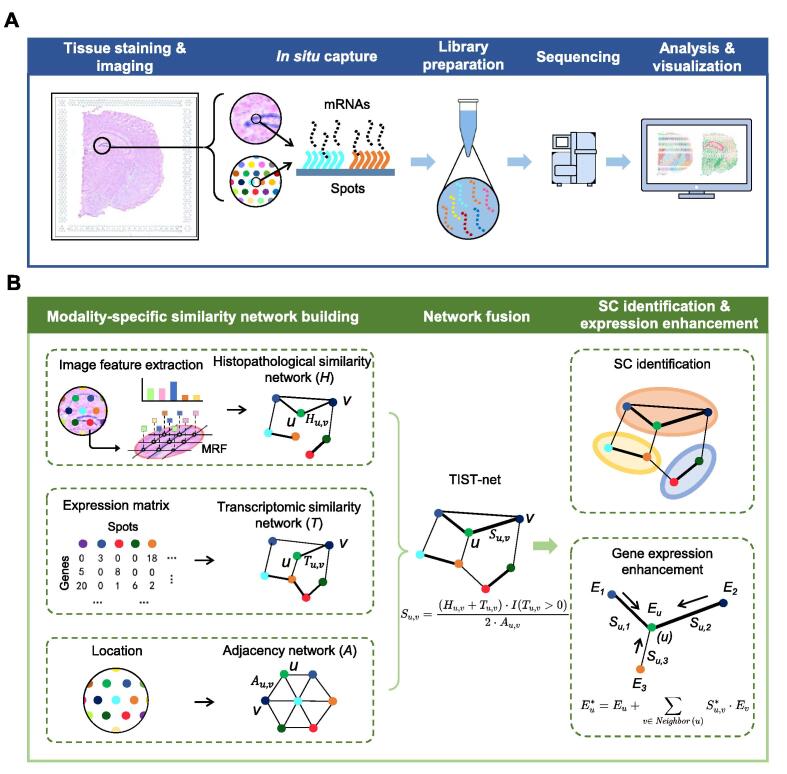
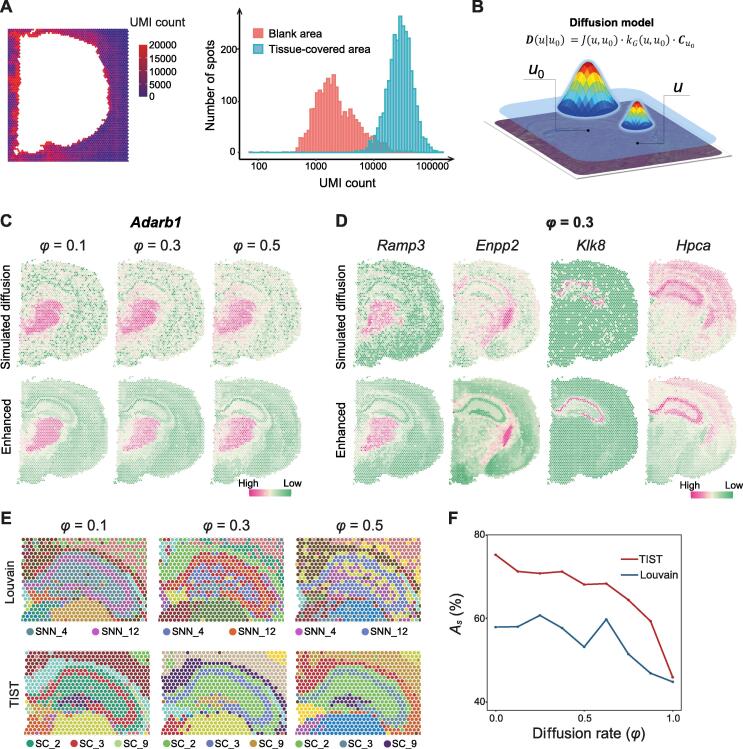
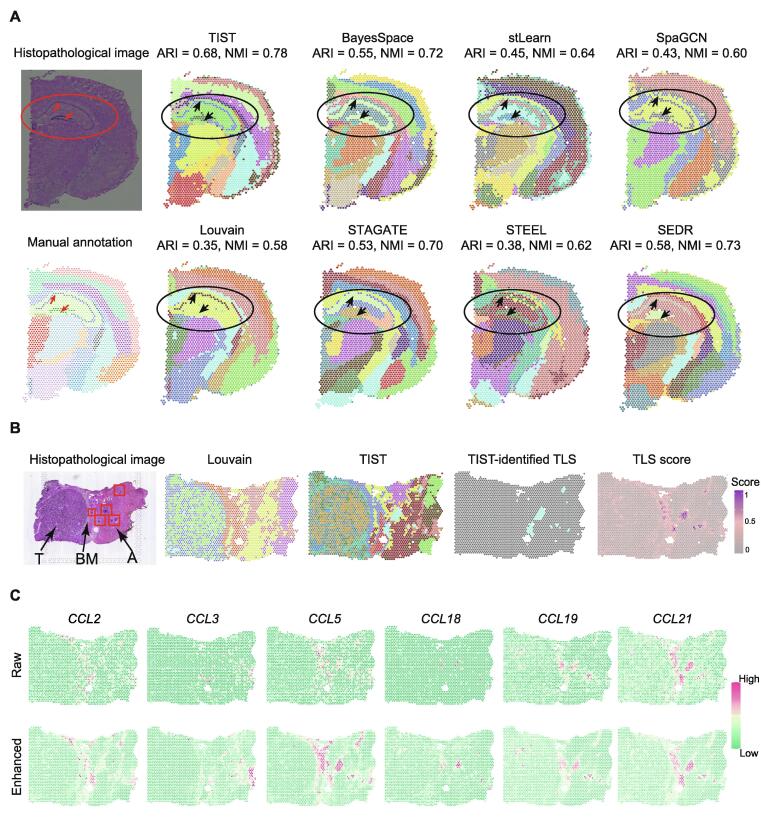
Sequencing-based spatial transcriptomics (ST) is an emerging technology to study in situ gene expression patterns at the whole-genome scale. Currently, ST data analysis is still complicated by high technical noises and low resolution. In addition to the transcriptomic data, matched histopathological images are usually generated for the same tissue sample along the ST experiment. The matched high-resolution histopathological images provide complementary cellular phenotypical information, providing an opportunity to mitigate the noises in ST data. We present a novel ST data analysis method called transcriptome and histopathological image integrative analysis for ST (TIST), which enables the identification of spatial clusters (SCs) and the enhancement of spatial gene expression patterns by integrative analysis of matched transcriptomic data and images. TIST devises a histopathological feature extraction method based on Markov random field (MRF) to learn the cellular features from histopathological images, and integrates them with the transcriptomic data and location information as a network, termed TIST-net. Based on TIST-net, SCs are identified by a random walk-based strategy, and gene expression patterns are enhanced by neighborhood smoothing. We benchmark TIST on both simulated datasets and 32 real samples against several state-of-the-art methods. Results show that TIST is robust to technical noises on multiple analysis tasks for sequencing-based ST data and can find interesting microstructures in different biological scenarios. TIST is available at http://lifeome.net/software/tist/ and https://ngdc.cncb.ac.cn/biocode/tools/BT007317.
期刊介绍:
Genomics, Proteomics and Bioinformatics (GPB) is the official journal of the Beijing Institute of Genomics, Chinese Academy of Sciences / China National Center for Bioinformation and Genetics Society of China. It aims to disseminate new developments in the field of omics and bioinformatics, publish high-quality discoveries quickly, and promote open access and online publication. GPB welcomes submissions in all areas of life science, biology, and biomedicine, with a focus on large data acquisition, analysis, and curation. Manuscripts covering omics and related bioinformatics topics are particularly encouraged. GPB is indexed/abstracted by PubMed/MEDLINE, PubMed Central, Scopus, BIOSIS Previews, Chemical Abstracts, CSCD, among others.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


