Sergio Carrera, Ana Belén Rodríguez-Martínez, Intza Garin, Esther Sarasola, Cristina Martínez, Hiart Maortua, Almudena Callejo, Abigail Ruiz de Lobera, Alberto Muñoz, Nagore Miñambres, Pablo Jiménez-Labaig
{"title":"在一个怀疑遗传性结直肠癌综合征的家族中首次报道了种系杂合外显子8-11致病性BARD1基因缺失:不仅仅是偶然发现?","authors":"Sergio Carrera, Ana Belén Rodríguez-Martínez, Intza Garin, Esther Sarasola, Cristina Martínez, Hiart Maortua, Almudena Callejo, Abigail Ruiz de Lobera, Alberto Muñoz, Nagore Miñambres, Pablo Jiménez-Labaig","doi":"10.1186/s13053-023-00246-4","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Colorectal cancer (CRC) is a highly prevalent disease in developed countries. Inherited Mendelian causes account for approximately 5% of CRC cases, with Lynch syndrome and familial adenomatous polyposis being the most prevalent forms. Scientific efforts are focused on the discovery of new candidate genes associated with CRC and new associations of phenotypes with well-established cancer-related genes. BRCA1-associated ring domain (BARD1) gene deleterious germline variants are associated with a moderate increase in the relative risk of breast cancer, but their association with other neoplasms, such as CRC, remains unclear.</p><p><strong>Case presentation: </strong>We present the case of a 49-year-old male diagnosed with rectal adenocarcinoma whose maternal family fulfilled Amsterdam clinical criteria for Lynch syndrome. Genetic test confirmed the presence in heterozygosis of a germline pathogenic deletion of exons 8-11 in BARD1 gene. The predictive genetic study of the family revealed the presence of this pathogenic variant in his deceased cancer affected relatives, confirming co-segregation of the deletion with the disease.</p><p><strong>Conclusions: </strong>To the best of our knowledge, this is the first published work in which this BARD1 deletion is detected in a family with familial colorectal cancer type X (FCCTX) syndrome, in which the clinical criteria for Lynch syndrome without alteration of the DNA mismatch repair (MMR) system are fulfilled. Whether this incidental germline finding is the cause of familial colorectal aggregation remains to be elucidated in scientific forums. Patients should be carefully assessed in specific cancer genetic counseling units to account for hypothetical casual findings in other genes, in principle unrelated to the initial clinical suspicion, but with potential impact on their health.</p>","PeriodicalId":55058,"journal":{"name":"Hereditary Cancer in Clinical Practice","volume":"21 1","pages":"2"},"PeriodicalIF":2.4000,"publicationDate":"2023-01-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9883939/pdf/","citationCount":"0","resultStr":"{\"title\":\"Germline heterozygous exons 8-11 pathogenic BARD1 gene deletion reported for the first time in a family with suspicion of a hereditary colorectal cancer syndrome: more than an incidental finding?\",\"authors\":\"Sergio Carrera, Ana Belén Rodríguez-Martínez, Intza Garin, Esther Sarasola, Cristina Martínez, Hiart Maortua, Almudena Callejo, Abigail Ruiz de Lobera, Alberto Muñoz, Nagore Miñambres, Pablo Jiménez-Labaig\",\"doi\":\"10.1186/s13053-023-00246-4\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Colorectal cancer (CRC) is a highly prevalent disease in developed countries. Inherited Mendelian causes account for approximately 5% of CRC cases, with Lynch syndrome and familial adenomatous polyposis being the most prevalent forms. Scientific efforts are focused on the discovery of new candidate genes associated with CRC and new associations of phenotypes with well-established cancer-related genes. BRCA1-associated ring domain (BARD1) gene deleterious germline variants are associated with a moderate increase in the relative risk of breast cancer, but their association with other neoplasms, such as CRC, remains unclear.</p><p><strong>Case presentation: </strong>We present the case of a 49-year-old male diagnosed with rectal adenocarcinoma whose maternal family fulfilled Amsterdam clinical criteria for Lynch syndrome. Genetic test confirmed the presence in heterozygosis of a germline pathogenic deletion of exons 8-11 in BARD1 gene. The predictive genetic study of the family revealed the presence of this pathogenic variant in his deceased cancer affected relatives, confirming co-segregation of the deletion with the disease.</p><p><strong>Conclusions: </strong>To the best of our knowledge, this is the first published work in which this BARD1 deletion is detected in a family with familial colorectal cancer type X (FCCTX) syndrome, in which the clinical criteria for Lynch syndrome without alteration of the DNA mismatch repair (MMR) system are fulfilled. Whether this incidental germline finding is the cause of familial colorectal aggregation remains to be elucidated in scientific forums. Patients should be carefully assessed in specific cancer genetic counseling units to account for hypothetical casual findings in other genes, in principle unrelated to the initial clinical suspicion, but with potential impact on their health.</p>\",\"PeriodicalId\":55058,\"journal\":{\"name\":\"Hereditary Cancer in Clinical Practice\",\"volume\":\"21 1\",\"pages\":\"2\"},\"PeriodicalIF\":2.4000,\"publicationDate\":\"2023-01-28\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9883939/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Hereditary Cancer in Clinical Practice\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1186/s13053-023-00246-4\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"ONCOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Hereditary Cancer in Clinical Practice","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s13053-023-00246-4","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"ONCOLOGY","Score":null,"Total":0}
Germline heterozygous exons 8-11 pathogenic BARD1 gene deletion reported for the first time in a family with suspicion of a hereditary colorectal cancer syndrome: more than an incidental finding?
Background: Colorectal cancer (CRC) is a highly prevalent disease in developed countries. Inherited Mendelian causes account for approximately 5% of CRC cases, with Lynch syndrome and familial adenomatous polyposis being the most prevalent forms. Scientific efforts are focused on the discovery of new candidate genes associated with CRC and new associations of phenotypes with well-established cancer-related genes. BRCA1-associated ring domain (BARD1) gene deleterious germline variants are associated with a moderate increase in the relative risk of breast cancer, but their association with other neoplasms, such as CRC, remains unclear.
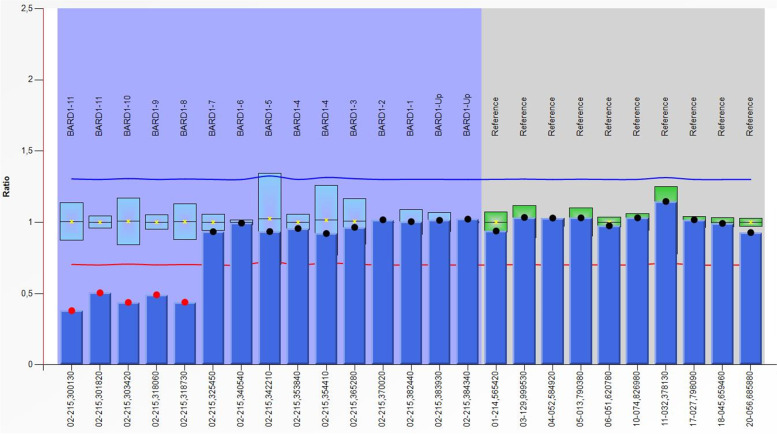
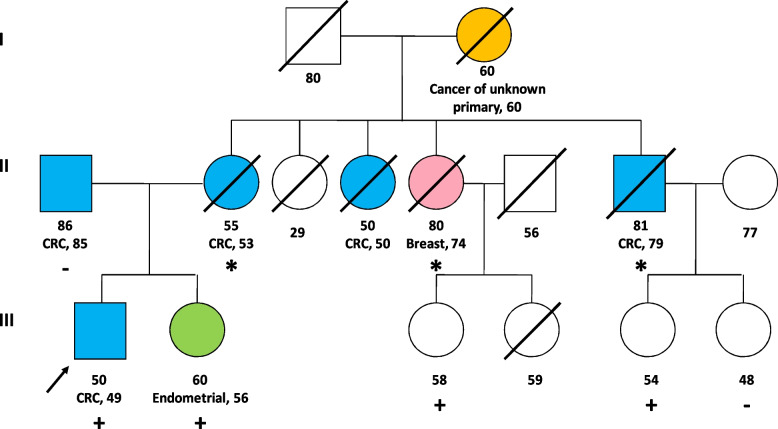
Case presentation: We present the case of a 49-year-old male diagnosed with rectal adenocarcinoma whose maternal family fulfilled Amsterdam clinical criteria for Lynch syndrome. Genetic test confirmed the presence in heterozygosis of a germline pathogenic deletion of exons 8-11 in BARD1 gene. The predictive genetic study of the family revealed the presence of this pathogenic variant in his deceased cancer affected relatives, confirming co-segregation of the deletion with the disease.
Conclusions: To the best of our knowledge, this is the first published work in which this BARD1 deletion is detected in a family with familial colorectal cancer type X (FCCTX) syndrome, in which the clinical criteria for Lynch syndrome without alteration of the DNA mismatch repair (MMR) system are fulfilled. Whether this incidental germline finding is the cause of familial colorectal aggregation remains to be elucidated in scientific forums. Patients should be carefully assessed in specific cancer genetic counseling units to account for hypothetical casual findings in other genes, in principle unrelated to the initial clinical suspicion, but with potential impact on their health.
期刊介绍:
Hereditary Cancer in Clinical Practice is an open access journal that publishes articles of interest for the cancer genetics community and serves as a discussion forum for the development appropriate healthcare strategies.
Cancer genetics encompasses a wide variety of disciplines and knowledge in the field is rapidly growing, especially as the amount of information linking genetic differences to inherited cancer predispositions continues expanding. With the increased knowledge of genetic variability and how this relates to cancer risk there is a growing demand not only to disseminate this information into clinical practice but also to enable competent debate concerning how such information is managed and what it implies for patient care.
Topics covered by the journal include but are not limited to:
Original research articles on any aspect of inherited predispositions to cancer.
Reviews of inherited cancer predispositions.
Application of molecular and cytogenetic analysis to clinical decision making.
Clinical aspects of the management of hereditary cancers.
Genetic counselling issues associated with cancer genetics.
The role of registries in improving health care of patients with an inherited predisposition to cancer.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


