{"title":"Epac活化抑制il -6诱导的心肌细胞功能障碍。","authors":"Huiling Jin, Takayuki Fujita, Meihua Jin, Reiko Kurotani, Yuko Hidaka, Wenqian Cai, Kenji Suita, Rajesh Prajapati, Chen Liang, Yoshiki Ohnuki, Yasumasa Mototani, Masanari Umemura, Utako Yokoyama, Motohiko Sato, Satoshi Okumura, Yoshihiro Ishikawa","doi":"10.1007/s12576-016-0509-5","DOIUrl":null,"url":null,"abstract":"<p><p>Pro-inflammatory cytokines are released in septic shock and impair cardiac function via the Jak-STAT pathway. It is well known that sympathetic and thus catecholamine signaling is activated thereafter to compensate for cardiac dysfunction. The mechanism of such compensation by catecholamine signaling has been traditionally understood to be cyclic AMP-dependent protein kinase (PKA)-mediated enforcement of cardiac contractility. We hypothesized that the exchange protein activated by cAMP (Epac), a newly identified target of cAMP signaling that functions independently of PKA, also plays a key role in this mechanism. In cultured cardiac myocytes, activation of Epac attenuated the inhibitory effect of interleukin-6 on the increase of intracellular Ca<sup>2+</sup> concentration and contractility in response to isoproterenol, most likely through inhibition of the Jak-STAT pathway via SOCS3, with subsequent changes in inducible nitric oxide synthase expression. These findings suggest a new role of catecholamine signaling in compensating for cardiac dysfunction in heart failure. Epac and its downstream pathway may be a novel target for treating cardiac dysfunction in endotoxemia.</p>","PeriodicalId":22836,"journal":{"name":"The Journal of Physiological Sciences","volume":"13 1","pages":"77-87"},"PeriodicalIF":0.0000,"publicationDate":"2018-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6353818/pdf/","citationCount":"0","resultStr":"{\"title\":\"Epac activation inhibits IL-6-induced cardiac myocyte dysfunction.\",\"authors\":\"Huiling Jin, Takayuki Fujita, Meihua Jin, Reiko Kurotani, Yuko Hidaka, Wenqian Cai, Kenji Suita, Rajesh Prajapati, Chen Liang, Yoshiki Ohnuki, Yasumasa Mototani, Masanari Umemura, Utako Yokoyama, Motohiko Sato, Satoshi Okumura, Yoshihiro Ishikawa\",\"doi\":\"10.1007/s12576-016-0509-5\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Pro-inflammatory cytokines are released in septic shock and impair cardiac function via the Jak-STAT pathway. It is well known that sympathetic and thus catecholamine signaling is activated thereafter to compensate for cardiac dysfunction. The mechanism of such compensation by catecholamine signaling has been traditionally understood to be cyclic AMP-dependent protein kinase (PKA)-mediated enforcement of cardiac contractility. We hypothesized that the exchange protein activated by cAMP (Epac), a newly identified target of cAMP signaling that functions independently of PKA, also plays a key role in this mechanism. In cultured cardiac myocytes, activation of Epac attenuated the inhibitory effect of interleukin-6 on the increase of intracellular Ca<sup>2+</sup> concentration and contractility in response to isoproterenol, most likely through inhibition of the Jak-STAT pathway via SOCS3, with subsequent changes in inducible nitric oxide synthase expression. These findings suggest a new role of catecholamine signaling in compensating for cardiac dysfunction in heart failure. Epac and its downstream pathway may be a novel target for treating cardiac dysfunction in endotoxemia.</p>\",\"PeriodicalId\":22836,\"journal\":{\"name\":\"The Journal of Physiological Sciences\",\"volume\":\"13 1\",\"pages\":\"77-87\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2018-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6353818/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Journal of Physiological Sciences\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1007/s12576-016-0509-5\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2016/12/19 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physiological Sciences","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1007/s12576-016-0509-5","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2016/12/19 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
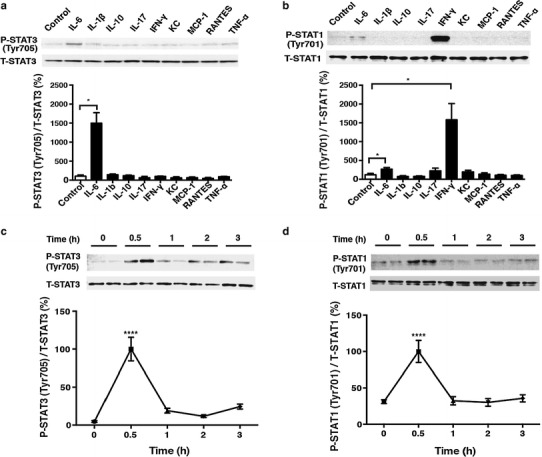
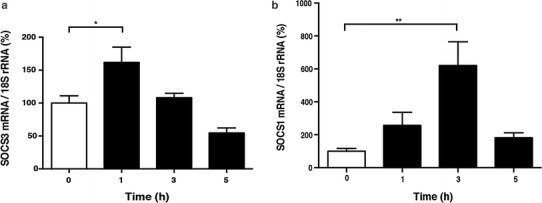
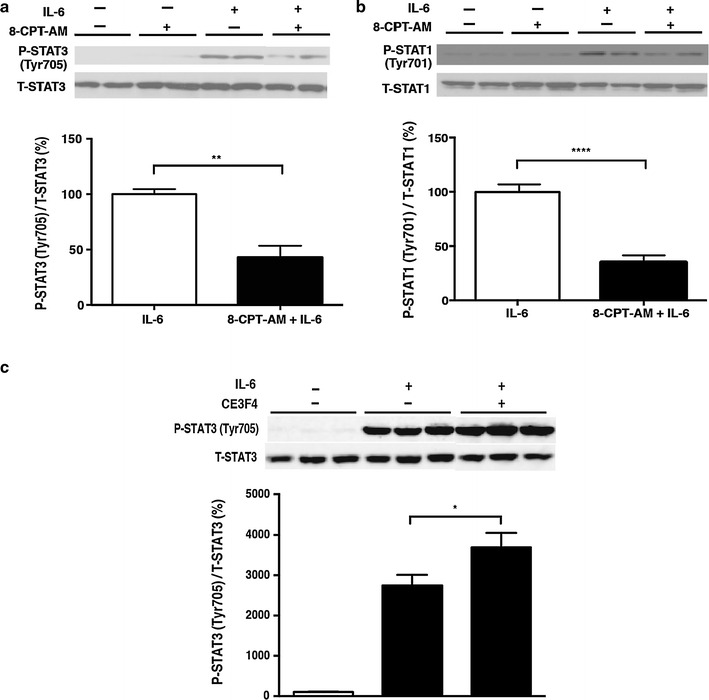
Pro-inflammatory cytokines are released in septic shock and impair cardiac function via the Jak-STAT pathway. It is well known that sympathetic and thus catecholamine signaling is activated thereafter to compensate for cardiac dysfunction. The mechanism of such compensation by catecholamine signaling has been traditionally understood to be cyclic AMP-dependent protein kinase (PKA)-mediated enforcement of cardiac contractility. We hypothesized that the exchange protein activated by cAMP (Epac), a newly identified target of cAMP signaling that functions independently of PKA, also plays a key role in this mechanism. In cultured cardiac myocytes, activation of Epac attenuated the inhibitory effect of interleukin-6 on the increase of intracellular Ca2+ concentration and contractility in response to isoproterenol, most likely through inhibition of the Jak-STAT pathway via SOCS3, with subsequent changes in inducible nitric oxide synthase expression. These findings suggest a new role of catecholamine signaling in compensating for cardiac dysfunction in heart failure. Epac and its downstream pathway may be a novel target for treating cardiac dysfunction in endotoxemia.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


