Gabriel Siqueira, Alexsandro Oliveira Alexandrino, Zanoni Dias
{"title":"考虑重复基因、基因间区域和indel的符号重排距离","authors":"Gabriel Siqueira, Alexsandro Oliveira Alexandrino, Zanoni Dias","doi":"10.1007/s10878-023-01083-w","DOIUrl":null,"url":null,"abstract":"<p>Genome rearrangement distance problems allow to estimate the evolutionary distance between genomes. These problems aim to compute the minimum number of mutations called rearrangement events necessary to transform one genome into another. Two commonly studied rearrangements are the reversal, which inverts a sequence of genes, and the transposition, which exchanges two consecutive sequences of genes. Seminal works on this topic focused on the sequence of genes and assumed that each gene occurs exactly once on each genome. More realistic models have been assuming that a gene may have multiple copies or may appear in only one of the genomes. Other models also take into account the nucleotides between consecutive pairs of genes, which are called intergenic regions. This work combines all these generalizations defining the signed intergenic reversal distance (SIRD), the signed intergenic reversal and transposition distance (SIRTD), the signed intergenic reversal and indels distance (SIRID), and the signed intergenic reversal, transposition, and indels distance (SIRTID) problems. We show a relation between these problems and the signed minimum common intergenic string partition (SMCISP) problem. From such relation, we derive <span>\\(\\varTheta (k)\\)</span>-approximation algorithms for the SIRD and the SIRTD problems, where <i>k</i> is maximum number of copies of a gene in the genomes. These algorithms also work as heuristics for the SIRID and SIRTID problems. Additionally, we present some parametrized algorithms for SMCISP that ensure constant approximation factors for the distance problems. Our experimental tests on simulated genomes show an improvement on the rearrangement distances with the use of the partition algorithms.\n</p>","PeriodicalId":50231,"journal":{"name":"Journal of Combinatorial Optimization","volume":null,"pages":null},"PeriodicalIF":0.9000,"publicationDate":"2023-09-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Signed rearrangement distances considering repeated genes, intergenic regions, and indels\",\"authors\":\"Gabriel Siqueira, Alexsandro Oliveira Alexandrino, Zanoni Dias\",\"doi\":\"10.1007/s10878-023-01083-w\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Genome rearrangement distance problems allow to estimate the evolutionary distance between genomes. These problems aim to compute the minimum number of mutations called rearrangement events necessary to transform one genome into another. Two commonly studied rearrangements are the reversal, which inverts a sequence of genes, and the transposition, which exchanges two consecutive sequences of genes. Seminal works on this topic focused on the sequence of genes and assumed that each gene occurs exactly once on each genome. More realistic models have been assuming that a gene may have multiple copies or may appear in only one of the genomes. Other models also take into account the nucleotides between consecutive pairs of genes, which are called intergenic regions. This work combines all these generalizations defining the signed intergenic reversal distance (SIRD), the signed intergenic reversal and transposition distance (SIRTD), the signed intergenic reversal and indels distance (SIRID), and the signed intergenic reversal, transposition, and indels distance (SIRTID) problems. We show a relation between these problems and the signed minimum common intergenic string partition (SMCISP) problem. From such relation, we derive <span>\\\\(\\\\varTheta (k)\\\\)</span>-approximation algorithms for the SIRD and the SIRTD problems, where <i>k</i> is maximum number of copies of a gene in the genomes. These algorithms also work as heuristics for the SIRID and SIRTID problems. Additionally, we present some parametrized algorithms for SMCISP that ensure constant approximation factors for the distance problems. Our experimental tests on simulated genomes show an improvement on the rearrangement distances with the use of the partition algorithms.\\n</p>\",\"PeriodicalId\":50231,\"journal\":{\"name\":\"Journal of Combinatorial Optimization\",\"volume\":null,\"pages\":null},\"PeriodicalIF\":0.9000,\"publicationDate\":\"2023-09-10\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Combinatorial Optimization\",\"FirstCategoryId\":\"100\",\"ListUrlMain\":\"https://doi.org/10.1007/s10878-023-01083-w\",\"RegionNum\":4,\"RegionCategory\":\"数学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"COMPUTER SCIENCE, INTERDISCIPLINARY APPLICATIONS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Combinatorial Optimization","FirstCategoryId":"100","ListUrlMain":"https://doi.org/10.1007/s10878-023-01083-w","RegionNum":4,"RegionCategory":"数学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"COMPUTER SCIENCE, INTERDISCIPLINARY APPLICATIONS","Score":null,"Total":0}
Signed rearrangement distances considering repeated genes, intergenic regions, and indels
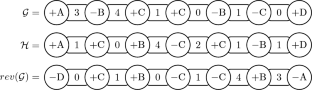
Genome rearrangement distance problems allow to estimate the evolutionary distance between genomes. These problems aim to compute the minimum number of mutations called rearrangement events necessary to transform one genome into another. Two commonly studied rearrangements are the reversal, which inverts a sequence of genes, and the transposition, which exchanges two consecutive sequences of genes. Seminal works on this topic focused on the sequence of genes and assumed that each gene occurs exactly once on each genome. More realistic models have been assuming that a gene may have multiple copies or may appear in only one of the genomes. Other models also take into account the nucleotides between consecutive pairs of genes, which are called intergenic regions. This work combines all these generalizations defining the signed intergenic reversal distance (SIRD), the signed intergenic reversal and transposition distance (SIRTD), the signed intergenic reversal and indels distance (SIRID), and the signed intergenic reversal, transposition, and indels distance (SIRTID) problems. We show a relation between these problems and the signed minimum common intergenic string partition (SMCISP) problem. From such relation, we derive \(\varTheta (k)\)-approximation algorithms for the SIRD and the SIRTD problems, where k is maximum number of copies of a gene in the genomes. These algorithms also work as heuristics for the SIRID and SIRTID problems. Additionally, we present some parametrized algorithms for SMCISP that ensure constant approximation factors for the distance problems. Our experimental tests on simulated genomes show an improvement on the rearrangement distances with the use of the partition algorithms.
期刊介绍:
The objective of Journal of Combinatorial Optimization is to advance and promote the theory and applications of combinatorial optimization, which is an area of research at the intersection of applied mathematics, computer science, and operations research and which overlaps with many other areas such as computation complexity, computational biology, VLSI design, communication networks, and management science. It includes complexity analysis and algorithm design for combinatorial optimization problems, numerical experiments and problem discovery with applications in science and engineering.
The Journal of Combinatorial Optimization publishes refereed papers dealing with all theoretical, computational and applied aspects of combinatorial optimization. It also publishes reviews of appropriate books and special issues of journals.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


