{"title":"杂合子X连锁Alport综合征的散发病例。","authors":"Jonathan E Zuckerman, Rachana Srivastava","doi":"10.1159/000530994","DOIUrl":null,"url":null,"abstract":"<p><p>Alport syndrome is a genetically and phenotypically heterogeneous disorder that can be transmitted in an X-linked, autosomal recessive, or autosomal dominant fashion and can affect glomerular, cochlear, and ocular basement membranes. The disorder results from mutations in the collagen IV genes <i>COL4A5</i> (X chromosome), <i>COL4A3</i>, and <i>COL4A4</i>. Alport patients are at lifetime risk for kidney failure, sensorineural deafness, and ocular abnormalities. Males with Alport syndrome typically present with severe phenotype with progression to end-stage kidney disease and/or sensorineural deafness and eye changes. Females generally having less severe presentation and diagnosis of X-linked Alport syndrome are generally not considered. Here, we report a case of a 3-year-old girl with gross hematuria, proteinuria, and chronic kidney disease who was found to have features of Alport syndrome on kidney biopsy and a sporadic heterozygous pathogenic <i>COL4A5</i> deletion on molecular testing. This case report emphasizes the importance of kidney biopsy and molecular testing in the work up of pediatric patients with hematuria, proteinuria, and/or chronic kidney disease. It is also a poignant illustration that females with heterozygous X-linked <i>COL4A5</i> mutations are often affected patients. It further illustrates the phenomenon of sporadic occurrence of genetic kidney disease in the absence of family history of kidney disease.</p>","PeriodicalId":73177,"journal":{"name":"Glomerular diseases","volume":"3 1","pages":"126-131"},"PeriodicalIF":0.0000,"publicationDate":"2023-05-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10601899/pdf/","citationCount":"0","resultStr":"{\"title\":\"Sporadic Case of Heterozygous X-Linked Alport Syndrome.\",\"authors\":\"Jonathan E Zuckerman, Rachana Srivastava\",\"doi\":\"10.1159/000530994\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Alport syndrome is a genetically and phenotypically heterogeneous disorder that can be transmitted in an X-linked, autosomal recessive, or autosomal dominant fashion and can affect glomerular, cochlear, and ocular basement membranes. The disorder results from mutations in the collagen IV genes <i>COL4A5</i> (X chromosome), <i>COL4A3</i>, and <i>COL4A4</i>. Alport patients are at lifetime risk for kidney failure, sensorineural deafness, and ocular abnormalities. Males with Alport syndrome typically present with severe phenotype with progression to end-stage kidney disease and/or sensorineural deafness and eye changes. Females generally having less severe presentation and diagnosis of X-linked Alport syndrome are generally not considered. Here, we report a case of a 3-year-old girl with gross hematuria, proteinuria, and chronic kidney disease who was found to have features of Alport syndrome on kidney biopsy and a sporadic heterozygous pathogenic <i>COL4A5</i> deletion on molecular testing. This case report emphasizes the importance of kidney biopsy and molecular testing in the work up of pediatric patients with hematuria, proteinuria, and/or chronic kidney disease. It is also a poignant illustration that females with heterozygous X-linked <i>COL4A5</i> mutations are often affected patients. It further illustrates the phenomenon of sporadic occurrence of genetic kidney disease in the absence of family history of kidney disease.</p>\",\"PeriodicalId\":73177,\"journal\":{\"name\":\"Glomerular diseases\",\"volume\":\"3 1\",\"pages\":\"126-131\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2023-05-16\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10601899/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Glomerular diseases\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1159/000530994\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Glomerular diseases","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1159/000530994","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
Sporadic Case of Heterozygous X-Linked Alport Syndrome.
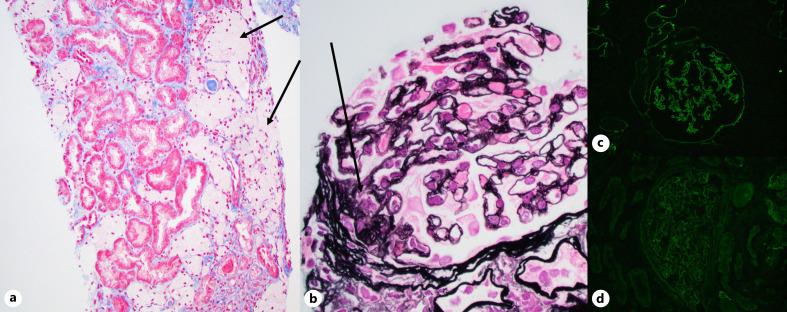
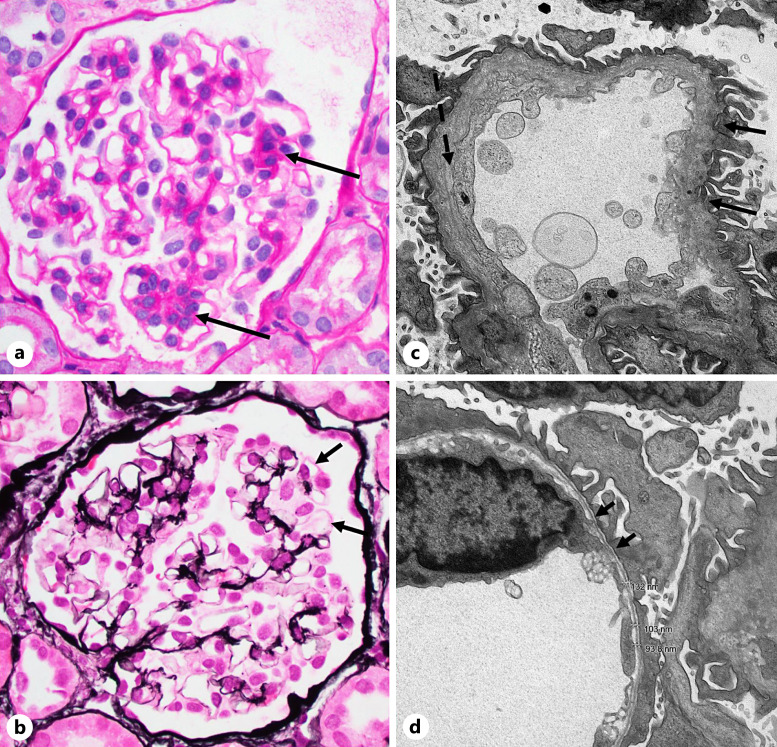
Alport syndrome is a genetically and phenotypically heterogeneous disorder that can be transmitted in an X-linked, autosomal recessive, or autosomal dominant fashion and can affect glomerular, cochlear, and ocular basement membranes. The disorder results from mutations in the collagen IV genes COL4A5 (X chromosome), COL4A3, and COL4A4. Alport patients are at lifetime risk for kidney failure, sensorineural deafness, and ocular abnormalities. Males with Alport syndrome typically present with severe phenotype with progression to end-stage kidney disease and/or sensorineural deafness and eye changes. Females generally having less severe presentation and diagnosis of X-linked Alport syndrome are generally not considered. Here, we report a case of a 3-year-old girl with gross hematuria, proteinuria, and chronic kidney disease who was found to have features of Alport syndrome on kidney biopsy and a sporadic heterozygous pathogenic COL4A5 deletion on molecular testing. This case report emphasizes the importance of kidney biopsy and molecular testing in the work up of pediatric patients with hematuria, proteinuria, and/or chronic kidney disease. It is also a poignant illustration that females with heterozygous X-linked COL4A5 mutations are often affected patients. It further illustrates the phenomenon of sporadic occurrence of genetic kidney disease in the absence of family history of kidney disease.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


