{"title":"法布里病表现为终末期肾病。","authors":"Madeleine V Pahl, Jean Hou","doi":"10.1159/000533502","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Fabry disease (FD) is an X-linked disorder due to a pathogenic variant of the <i>GLA</i> gene that codes for the alpha-galactosidase enzyme. The reduced or absent activity of the enzyme results in lysosomal accumulation of globotriosylceramide and its derivative, globotriaosylsphingosine, in a variety of cells, leading to a variety of complications including cardiac, renal, and cerebrovascular disorders. Early diagnosis is critically important for the selection of therapeutic treatments, which are essential for improving outcomes. Here we present a case of FD diagnosed at the time of end-stage kidney disease presentation.</p><p><strong>Summary: </strong>A 40-year-old man with a history of seizures presented with increased serum creatinine, nephrotic rage proteinuria, and new-onset hypertension. A renal biopsy revealed numerous, whorled, and lamellated cytoplasmic inclusions in podocytes, glomerular peritubular capillary endothelial cells, mesangial cells, arterial myocytes, and interstitial macrophages. Ultrastructural analysis confirmed the presence of glycosphingolipid inclusions and enlarged lysosomes packed with multi-lamellated structures (\"zebra\" bodies). The findings were suggestive of a lysosomal storage disorder, and testing for alpha-galactosidase A levels revealed near-absent enzyme activity, confirming the diagnosis of advanced FD.</p><p><strong>Key messages: </strong>The diagnosis of FD can be challenging as the manifestations of the disease are nonspecific, and patients can present early with classical symptoms or late with non-classical patterns of involvement. We will discuss strategies to identify the disorder early by reviewing the classical and non-classical presentations and further outline currently available and potential future treatment options.</p>","PeriodicalId":73177,"journal":{"name":"Glomerular diseases","volume":"3 1","pages":"189-196"},"PeriodicalIF":0.0000,"publicationDate":"2023-08-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10601945/pdf/","citationCount":"0","resultStr":"{\"title\":\"Fabry Disease Presenting as End-Stage Kidney Disease.\",\"authors\":\"Madeleine V Pahl, Jean Hou\",\"doi\":\"10.1159/000533502\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Fabry disease (FD) is an X-linked disorder due to a pathogenic variant of the <i>GLA</i> gene that codes for the alpha-galactosidase enzyme. The reduced or absent activity of the enzyme results in lysosomal accumulation of globotriosylceramide and its derivative, globotriaosylsphingosine, in a variety of cells, leading to a variety of complications including cardiac, renal, and cerebrovascular disorders. Early diagnosis is critically important for the selection of therapeutic treatments, which are essential for improving outcomes. Here we present a case of FD diagnosed at the time of end-stage kidney disease presentation.</p><p><strong>Summary: </strong>A 40-year-old man with a history of seizures presented with increased serum creatinine, nephrotic rage proteinuria, and new-onset hypertension. A renal biopsy revealed numerous, whorled, and lamellated cytoplasmic inclusions in podocytes, glomerular peritubular capillary endothelial cells, mesangial cells, arterial myocytes, and interstitial macrophages. Ultrastructural analysis confirmed the presence of glycosphingolipid inclusions and enlarged lysosomes packed with multi-lamellated structures (\\\"zebra\\\" bodies). The findings were suggestive of a lysosomal storage disorder, and testing for alpha-galactosidase A levels revealed near-absent enzyme activity, confirming the diagnosis of advanced FD.</p><p><strong>Key messages: </strong>The diagnosis of FD can be challenging as the manifestations of the disease are nonspecific, and patients can present early with classical symptoms or late with non-classical patterns of involvement. We will discuss strategies to identify the disorder early by reviewing the classical and non-classical presentations and further outline currently available and potential future treatment options.</p>\",\"PeriodicalId\":73177,\"journal\":{\"name\":\"Glomerular diseases\",\"volume\":\"3 1\",\"pages\":\"189-196\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2023-08-16\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10601945/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Glomerular diseases\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1159/000533502\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Glomerular diseases","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1159/000533502","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
Fabry Disease Presenting as End-Stage Kidney Disease.
Background: Fabry disease (FD) is an X-linked disorder due to a pathogenic variant of the GLA gene that codes for the alpha-galactosidase enzyme. The reduced or absent activity of the enzyme results in lysosomal accumulation of globotriosylceramide and its derivative, globotriaosylsphingosine, in a variety of cells, leading to a variety of complications including cardiac, renal, and cerebrovascular disorders. Early diagnosis is critically important for the selection of therapeutic treatments, which are essential for improving outcomes. Here we present a case of FD diagnosed at the time of end-stage kidney disease presentation.
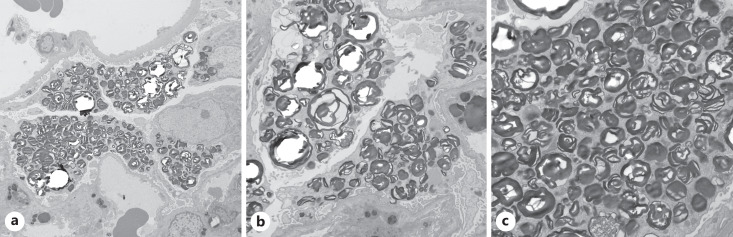
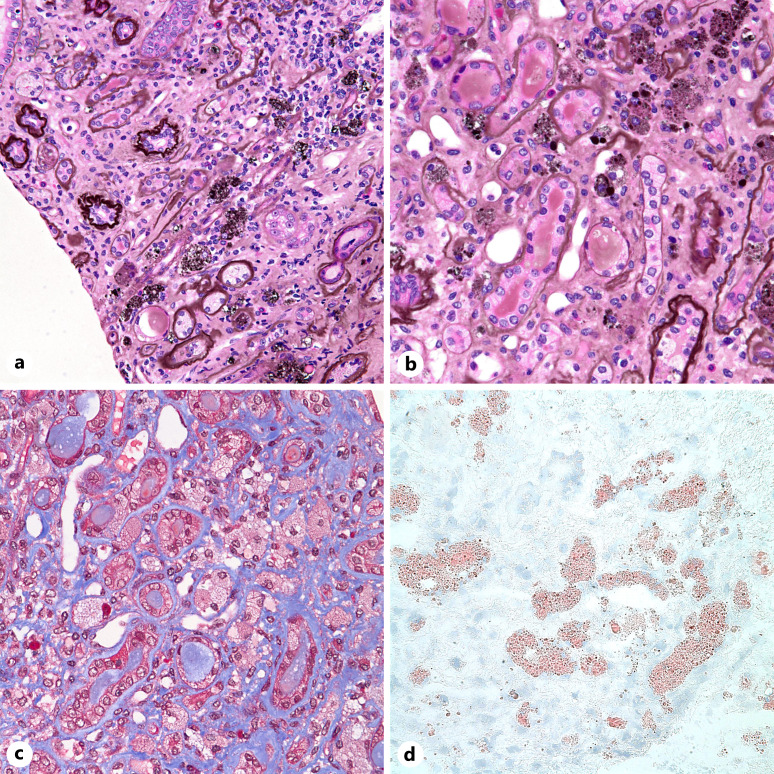
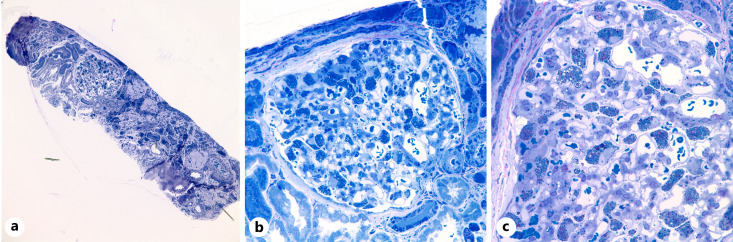
Summary: A 40-year-old man with a history of seizures presented with increased serum creatinine, nephrotic rage proteinuria, and new-onset hypertension. A renal biopsy revealed numerous, whorled, and lamellated cytoplasmic inclusions in podocytes, glomerular peritubular capillary endothelial cells, mesangial cells, arterial myocytes, and interstitial macrophages. Ultrastructural analysis confirmed the presence of glycosphingolipid inclusions and enlarged lysosomes packed with multi-lamellated structures ("zebra" bodies). The findings were suggestive of a lysosomal storage disorder, and testing for alpha-galactosidase A levels revealed near-absent enzyme activity, confirming the diagnosis of advanced FD.
Key messages: The diagnosis of FD can be challenging as the manifestations of the disease are nonspecific, and patients can present early with classical symptoms or late with non-classical patterns of involvement. We will discuss strategies to identify the disorder early by reviewing the classical and non-classical presentations and further outline currently available and potential future treatment options.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


