Matthew D Rannals, Stephanie Cerceo Page, Morganne N Campbell, Ryan A Gallo, Brent Mayfield, Brady J Maher
{"title":"转录因子 4 缺乏症的神经发育模型将共同离子通道作为皮特-霍普金斯综合症的潜在治疗靶点。","authors":"Matthew D Rannals, Stephanie Cerceo Page, Morganne N Campbell, Ryan A Gallo, Brent Mayfield, Brady J Maher","doi":"10.1080/21675511.2016.1220468","DOIUrl":null,"url":null,"abstract":"<p><p>The clinically pleiotropic gene, Transcription Factor 4 (TCF4), is a broadly expressed basic helix-loop-helix (bHLH) transcription factor linked to multiple neurodevelopmental disorders, including schizophrenia, 18q deletion syndrome, and Pitt Hopkins syndrome (PTHS). <i>In vivo</i> suppression of <i>Tcf4</i> by shRNA or mutation by CRISPR/Cas9 in the developing rat prefrontal cortex resulted in attenuated action potential output. To explain this intrinsic excitability deficit, we demonstrated that haploinsufficiency of TCF4 lead to the ectopic expression of two ion channels, <i>Scn10a</i> and <i>Kcnq1</i>. These targets of TCF4 regulation were identified through molecular profiling experiments that used translating ribosome affinity purification to enrich mRNA from genetically manipulated neurons. Using a mouse model of PTHS (<i>Tcf4</i><sup>+/tr</sup>), we observed a similar intrinsic excitability deficit, however the underlying mechanism appeared slightly different than our rat model - as <i>Scn10a</i> expression was similarly increased but <i>Kcnq1</i> expression was decreased. Here, we show that the truncated TCF4 protein expressed in our PTHS mouse model binds to wild-type TCF4 protein, and we suggest the difference in <i>Kcnq1</i> expression levels between these two rodent models appears to be explained by a dominant-negative function of the truncated TCF4 protein. Despite the differences in the underlying molecular mechanisms, we observed common underlying intrinsic excitability deficits that are consistent with ectopic expression of <i>Scn10a</i>. The converging molecular function of TCF4 across two independent rodent models indicates SCN10a is a potential therapeutic target for Pitt-Hopkins syndrome.</p>","PeriodicalId":74639,"journal":{"name":"Rare diseases (Austin, Tex.)","volume":"4 1","pages":"e1220468"},"PeriodicalIF":0.0000,"publicationDate":"2016-08-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5154382/pdf/","citationCount":"0","resultStr":"{\"title\":\"Neurodevelopmental models of transcription factor 4 deficiency converge on a common ion channel as a potential therapeutic target for Pitt Hopkins syndrome.\",\"authors\":\"Matthew D Rannals, Stephanie Cerceo Page, Morganne N Campbell, Ryan A Gallo, Brent Mayfield, Brady J Maher\",\"doi\":\"10.1080/21675511.2016.1220468\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>The clinically pleiotropic gene, Transcription Factor 4 (TCF4), is a broadly expressed basic helix-loop-helix (bHLH) transcription factor linked to multiple neurodevelopmental disorders, including schizophrenia, 18q deletion syndrome, and Pitt Hopkins syndrome (PTHS). <i>In vivo</i> suppression of <i>Tcf4</i> by shRNA or mutation by CRISPR/Cas9 in the developing rat prefrontal cortex resulted in attenuated action potential output. To explain this intrinsic excitability deficit, we demonstrated that haploinsufficiency of TCF4 lead to the ectopic expression of two ion channels, <i>Scn10a</i> and <i>Kcnq1</i>. These targets of TCF4 regulation were identified through molecular profiling experiments that used translating ribosome affinity purification to enrich mRNA from genetically manipulated neurons. Using a mouse model of PTHS (<i>Tcf4</i><sup>+/tr</sup>), we observed a similar intrinsic excitability deficit, however the underlying mechanism appeared slightly different than our rat model - as <i>Scn10a</i> expression was similarly increased but <i>Kcnq1</i> expression was decreased. Here, we show that the truncated TCF4 protein expressed in our PTHS mouse model binds to wild-type TCF4 protein, and we suggest the difference in <i>Kcnq1</i> expression levels between these two rodent models appears to be explained by a dominant-negative function of the truncated TCF4 protein. Despite the differences in the underlying molecular mechanisms, we observed common underlying intrinsic excitability deficits that are consistent with ectopic expression of <i>Scn10a</i>. The converging molecular function of TCF4 across two independent rodent models indicates SCN10a is a potential therapeutic target for Pitt-Hopkins syndrome.</p>\",\"PeriodicalId\":74639,\"journal\":{\"name\":\"Rare diseases (Austin, Tex.)\",\"volume\":\"4 1\",\"pages\":\"e1220468\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2016-08-05\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5154382/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Rare diseases (Austin, Tex.)\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1080/21675511.2016.1220468\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2016/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Rare diseases (Austin, Tex.)","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1080/21675511.2016.1220468","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2016/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
Neurodevelopmental models of transcription factor 4 deficiency converge on a common ion channel as a potential therapeutic target for Pitt Hopkins syndrome.
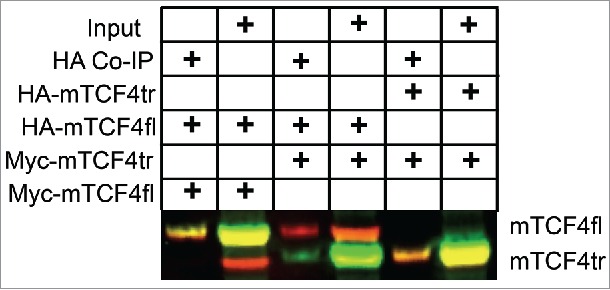
The clinically pleiotropic gene, Transcription Factor 4 (TCF4), is a broadly expressed basic helix-loop-helix (bHLH) transcription factor linked to multiple neurodevelopmental disorders, including schizophrenia, 18q deletion syndrome, and Pitt Hopkins syndrome (PTHS). In vivo suppression of Tcf4 by shRNA or mutation by CRISPR/Cas9 in the developing rat prefrontal cortex resulted in attenuated action potential output. To explain this intrinsic excitability deficit, we demonstrated that haploinsufficiency of TCF4 lead to the ectopic expression of two ion channels, Scn10a and Kcnq1. These targets of TCF4 regulation were identified through molecular profiling experiments that used translating ribosome affinity purification to enrich mRNA from genetically manipulated neurons. Using a mouse model of PTHS (Tcf4+/tr), we observed a similar intrinsic excitability deficit, however the underlying mechanism appeared slightly different than our rat model - as Scn10a expression was similarly increased but Kcnq1 expression was decreased. Here, we show that the truncated TCF4 protein expressed in our PTHS mouse model binds to wild-type TCF4 protein, and we suggest the difference in Kcnq1 expression levels between these two rodent models appears to be explained by a dominant-negative function of the truncated TCF4 protein. Despite the differences in the underlying molecular mechanisms, we observed common underlying intrinsic excitability deficits that are consistent with ectopic expression of Scn10a. The converging molecular function of TCF4 across two independent rodent models indicates SCN10a is a potential therapeutic target for Pitt-Hopkins syndrome.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


