Kyle R. Bryenton, Adebayo A. Adeleke, Stephen G. Dale, Erin R. Johnson
{"title":"离域误差:密度泛函理论中最大的突出挑战","authors":"Kyle R. Bryenton, Adebayo A. Adeleke, Stephen G. Dale, Erin R. Johnson","doi":"10.1002/wcms.1631","DOIUrl":null,"url":null,"abstract":"<p>Every day, density-functional theory (DFT) is routinely applied to computational modeling of molecules and materials with the expectation of high accuracy. However, in certain situations, popular density-functional approximations (DFAs) have the potential to give substantial quantitative, and even qualitative, errors. The most common class of error is delocalization error, which is an overarching term that also encompasses the one-electron self-interaction error. In our opinion, its resolution remains the greatest outstanding challenge in DFT development. In this paper, we review the history of delocalization error and provide several complimentary conceptual pictures for its interpretation, along with illustrative examples of its various manifestations. Approaches to reduce delocalization error are discussed, as is its interplay with other shortcomings of popular DFAs, including treatment of non-bonded repulsion and neglect of London dispersion.</p><p>This article is categorized under:\n </p>","PeriodicalId":236,"journal":{"name":"Wiley Interdisciplinary Reviews: Computational Molecular Science","volume":"13 2","pages":""},"PeriodicalIF":16.8000,"publicationDate":"2022-07-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"36","resultStr":"{\"title\":\"Delocalization error: The greatest outstanding challenge in density-functional theory\",\"authors\":\"Kyle R. Bryenton, Adebayo A. Adeleke, Stephen G. Dale, Erin R. Johnson\",\"doi\":\"10.1002/wcms.1631\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Every day, density-functional theory (DFT) is routinely applied to computational modeling of molecules and materials with the expectation of high accuracy. However, in certain situations, popular density-functional approximations (DFAs) have the potential to give substantial quantitative, and even qualitative, errors. The most common class of error is delocalization error, which is an overarching term that also encompasses the one-electron self-interaction error. In our opinion, its resolution remains the greatest outstanding challenge in DFT development. In this paper, we review the history of delocalization error and provide several complimentary conceptual pictures for its interpretation, along with illustrative examples of its various manifestations. Approaches to reduce delocalization error are discussed, as is its interplay with other shortcomings of popular DFAs, including treatment of non-bonded repulsion and neglect of London dispersion.</p><p>This article is categorized under:\\n </p>\",\"PeriodicalId\":236,\"journal\":{\"name\":\"Wiley Interdisciplinary Reviews: Computational Molecular Science\",\"volume\":\"13 2\",\"pages\":\"\"},\"PeriodicalIF\":16.8000,\"publicationDate\":\"2022-07-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"36\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Wiley Interdisciplinary Reviews: Computational Molecular Science\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/wcms.1631\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Wiley Interdisciplinary Reviews: Computational Molecular Science","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/wcms.1631","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
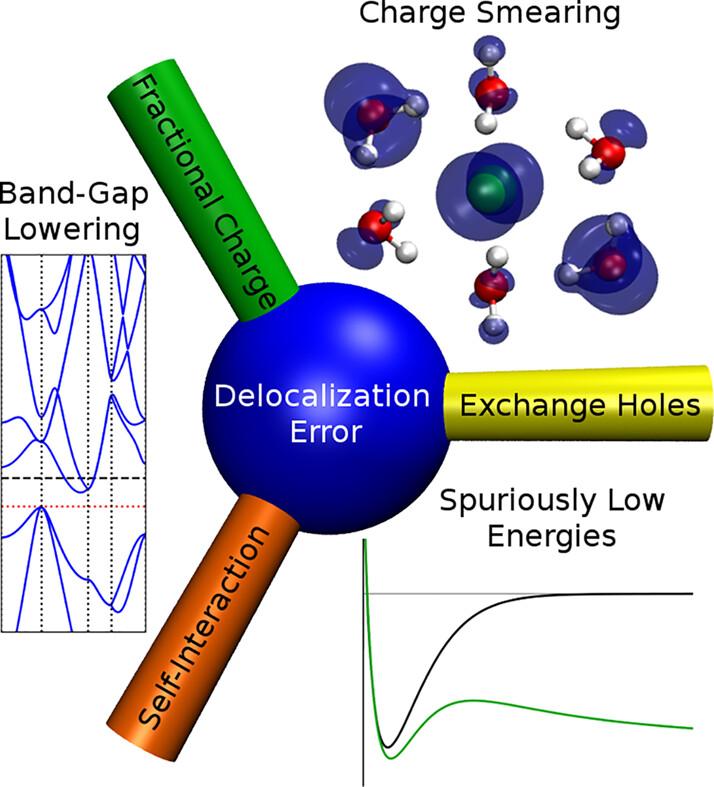
Delocalization error: The greatest outstanding challenge in density-functional theory
Every day, density-functional theory (DFT) is routinely applied to computational modeling of molecules and materials with the expectation of high accuracy. However, in certain situations, popular density-functional approximations (DFAs) have the potential to give substantial quantitative, and even qualitative, errors. The most common class of error is delocalization error, which is an overarching term that also encompasses the one-electron self-interaction error. In our opinion, its resolution remains the greatest outstanding challenge in DFT development. In this paper, we review the history of delocalization error and provide several complimentary conceptual pictures for its interpretation, along with illustrative examples of its various manifestations. Approaches to reduce delocalization error are discussed, as is its interplay with other shortcomings of popular DFAs, including treatment of non-bonded repulsion and neglect of London dispersion.
期刊介绍:
Computational molecular sciences harness the power of rigorous chemical and physical theories, employing computer-based modeling, specialized hardware, software development, algorithm design, and database management to explore and illuminate every facet of molecular sciences. These interdisciplinary approaches form a bridge between chemistry, biology, and materials sciences, establishing connections with adjacent application-driven fields in both chemistry and biology. WIREs Computational Molecular Science stands as a platform to comprehensively review and spotlight research from these dynamic and interconnected fields.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


