Stefan Vuckovic, Augusto Gerolin, Timothy J. Daas, Hilke Bahmann, Gero Friesecke, Paola Gori-Giorgi
{"title":"基于DFT强相互作用极限数学结构的密度泛函","authors":"Stefan Vuckovic, Augusto Gerolin, Timothy J. Daas, Hilke Bahmann, Gero Friesecke, Paola Gori-Giorgi","doi":"10.1002/wcms.1634","DOIUrl":null,"url":null,"abstract":"<p>While in principle exact, Kohn–Sham density functional theory—the workhorse of computational chemistry—must rely on approximations for the exchange–correlation functional. Despite staggering successes, present-day approximations still struggle when the effects of electron–electron correlation play a prominent role. The limit in which the electronic Coulomb repulsion completely dominates the exchange–correlation functional offers a well-defined mathematical framework that provides insight for new approximations able to deal with strong correlation. In particular, the mathematical structure of this limit, which is now well-established thanks to its reformulation as an optimal transport problem, points to the use of very different ingredients (or features) with respect to the traditional ones used in present approximations. We focus on strategies to use these new ingredients to build approximations for computational chemistry and highlight future promising directions.</p><p>This article is categorized under:\n </p>","PeriodicalId":236,"journal":{"name":"Wiley Interdisciplinary Reviews: Computational Molecular Science","volume":"13 2","pages":""},"PeriodicalIF":16.8000,"publicationDate":"2022-08-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/wcms.1634","citationCount":"13","resultStr":"{\"title\":\"Density functionals based on the mathematical structure of the strong-interaction limit of DFT\",\"authors\":\"Stefan Vuckovic, Augusto Gerolin, Timothy J. Daas, Hilke Bahmann, Gero Friesecke, Paola Gori-Giorgi\",\"doi\":\"10.1002/wcms.1634\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>While in principle exact, Kohn–Sham density functional theory—the workhorse of computational chemistry—must rely on approximations for the exchange–correlation functional. Despite staggering successes, present-day approximations still struggle when the effects of electron–electron correlation play a prominent role. The limit in which the electronic Coulomb repulsion completely dominates the exchange–correlation functional offers a well-defined mathematical framework that provides insight for new approximations able to deal with strong correlation. In particular, the mathematical structure of this limit, which is now well-established thanks to its reformulation as an optimal transport problem, points to the use of very different ingredients (or features) with respect to the traditional ones used in present approximations. We focus on strategies to use these new ingredients to build approximations for computational chemistry and highlight future promising directions.</p><p>This article is categorized under:\\n </p>\",\"PeriodicalId\":236,\"journal\":{\"name\":\"Wiley Interdisciplinary Reviews: Computational Molecular Science\",\"volume\":\"13 2\",\"pages\":\"\"},\"PeriodicalIF\":16.8000,\"publicationDate\":\"2022-08-29\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/wcms.1634\",\"citationCount\":\"13\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Wiley Interdisciplinary Reviews: Computational Molecular Science\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/wcms.1634\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Wiley Interdisciplinary Reviews: Computational Molecular Science","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/wcms.1634","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
Density functionals based on the mathematical structure of the strong-interaction limit of DFT
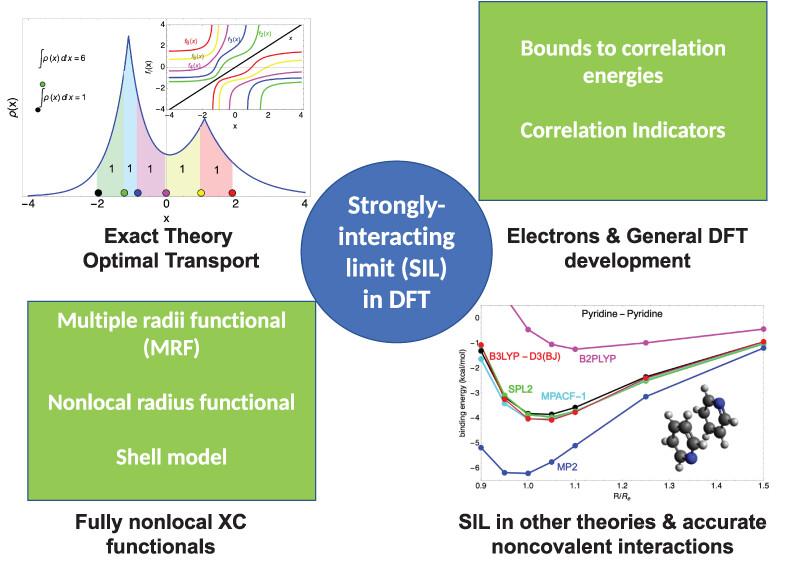
While in principle exact, Kohn–Sham density functional theory—the workhorse of computational chemistry—must rely on approximations for the exchange–correlation functional. Despite staggering successes, present-day approximations still struggle when the effects of electron–electron correlation play a prominent role. The limit in which the electronic Coulomb repulsion completely dominates the exchange–correlation functional offers a well-defined mathematical framework that provides insight for new approximations able to deal with strong correlation. In particular, the mathematical structure of this limit, which is now well-established thanks to its reformulation as an optimal transport problem, points to the use of very different ingredients (or features) with respect to the traditional ones used in present approximations. We focus on strategies to use these new ingredients to build approximations for computational chemistry and highlight future promising directions.
期刊介绍:
Computational molecular sciences harness the power of rigorous chemical and physical theories, employing computer-based modeling, specialized hardware, software development, algorithm design, and database management to explore and illuminate every facet of molecular sciences. These interdisciplinary approaches form a bridge between chemistry, biology, and materials sciences, establishing connections with adjacent application-driven fields in both chemistry and biology. WIREs Computational Molecular Science stands as a platform to comprehensively review and spotlight research from these dynamic and interconnected fields.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


