{"title":"分子、凝聚相和界面系统化学动力学模拟的原子神经网络表征:效率、可表征性和泛化","authors":"Yaolong Zhang, Qidong Lin, Bin Jiang","doi":"10.1002/wcms.1645","DOIUrl":null,"url":null,"abstract":"<p>Machine learning techniques have been widely applied in many fields of chemistry, physics, biology, and materials science. One of the most fruitful applications is machine learning of the complicated multidimensional function of potential energy or related electronic properties from discrete quantum chemical data. In particular, substantial efforts have been dedicated to developing various atomistic neural network (AtNN) representations, which refer to a family of methods expressing the targeted physical quantity as a sum of atomic components represented by atomic NNs. This class of approaches not only fully preserves the physical symmetry of the system but also scales linearly with respect to the size of a system, enabling accurate and efficient chemical dynamics and spectroscopic simulations in complicated systems and even a number of variably sized systems across the phases. In this review, we discuss different strategies in developing highly efficient and representable AtNN potentials, and in generalizing these scalar AtNN models to learn vectorial and tensorial quantities with the correct rotational equivariance. We also review active learning algorithms to generate practical AtNN models and present selected examples of AtNN applications in gas-surface systems to demonstrate their capabilities of accurately representing both molecular systems and condensed phase systems. We conclude this review by pointing out remaining challenges for the further development of more reliable, transferable, and scalable AtNN representations in more application scenarios.</p><p>This article is categorized under:\n </p>","PeriodicalId":236,"journal":{"name":"Wiley Interdisciplinary Reviews: Computational Molecular Science","volume":"13 3","pages":""},"PeriodicalIF":27.0000,"publicationDate":"2022-11-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"10","resultStr":"{\"title\":\"Atomistic neural network representations for chemical dynamics simulations of molecular, condensed phase, and interfacial systems: Efficiency, representability, and generalization\",\"authors\":\"Yaolong Zhang, Qidong Lin, Bin Jiang\",\"doi\":\"10.1002/wcms.1645\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Machine learning techniques have been widely applied in many fields of chemistry, physics, biology, and materials science. One of the most fruitful applications is machine learning of the complicated multidimensional function of potential energy or related electronic properties from discrete quantum chemical data. In particular, substantial efforts have been dedicated to developing various atomistic neural network (AtNN) representations, which refer to a family of methods expressing the targeted physical quantity as a sum of atomic components represented by atomic NNs. This class of approaches not only fully preserves the physical symmetry of the system but also scales linearly with respect to the size of a system, enabling accurate and efficient chemical dynamics and spectroscopic simulations in complicated systems and even a number of variably sized systems across the phases. In this review, we discuss different strategies in developing highly efficient and representable AtNN potentials, and in generalizing these scalar AtNN models to learn vectorial and tensorial quantities with the correct rotational equivariance. We also review active learning algorithms to generate practical AtNN models and present selected examples of AtNN applications in gas-surface systems to demonstrate their capabilities of accurately representing both molecular systems and condensed phase systems. We conclude this review by pointing out remaining challenges for the further development of more reliable, transferable, and scalable AtNN representations in more application scenarios.</p><p>This article is categorized under:\\n </p>\",\"PeriodicalId\":236,\"journal\":{\"name\":\"Wiley Interdisciplinary Reviews: Computational Molecular Science\",\"volume\":\"13 3\",\"pages\":\"\"},\"PeriodicalIF\":27.0000,\"publicationDate\":\"2022-11-16\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"10\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Wiley Interdisciplinary Reviews: Computational Molecular Science\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/wcms.1645\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Wiley Interdisciplinary Reviews: Computational Molecular Science","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/wcms.1645","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
Atomistic neural network representations for chemical dynamics simulations of molecular, condensed phase, and interfacial systems: Efficiency, representability, and generalization
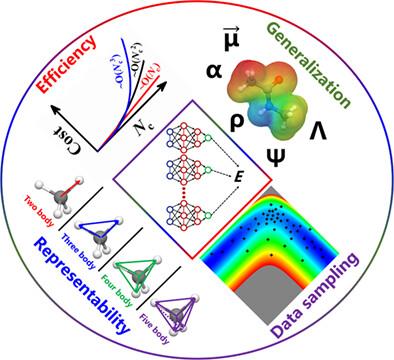
Machine learning techniques have been widely applied in many fields of chemistry, physics, biology, and materials science. One of the most fruitful applications is machine learning of the complicated multidimensional function of potential energy or related electronic properties from discrete quantum chemical data. In particular, substantial efforts have been dedicated to developing various atomistic neural network (AtNN) representations, which refer to a family of methods expressing the targeted physical quantity as a sum of atomic components represented by atomic NNs. This class of approaches not only fully preserves the physical symmetry of the system but also scales linearly with respect to the size of a system, enabling accurate and efficient chemical dynamics and spectroscopic simulations in complicated systems and even a number of variably sized systems across the phases. In this review, we discuss different strategies in developing highly efficient and representable AtNN potentials, and in generalizing these scalar AtNN models to learn vectorial and tensorial quantities with the correct rotational equivariance. We also review active learning algorithms to generate practical AtNN models and present selected examples of AtNN applications in gas-surface systems to demonstrate their capabilities of accurately representing both molecular systems and condensed phase systems. We conclude this review by pointing out remaining challenges for the further development of more reliable, transferable, and scalable AtNN representations in more application scenarios.
期刊介绍:
Computational molecular sciences harness the power of rigorous chemical and physical theories, employing computer-based modeling, specialized hardware, software development, algorithm design, and database management to explore and illuminate every facet of molecular sciences. These interdisciplinary approaches form a bridge between chemistry, biology, and materials sciences, establishing connections with adjacent application-driven fields in both chemistry and biology. WIREs Computational Molecular Science stands as a platform to comprehensively review and spotlight research from these dynamic and interconnected fields.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


