Qifeng Bai, Shuo Liu, Yanan Tian, Tingyang Xu, Antonio Jesús Banegas-Luna, Horacio Pérez-Sánchez, Junzhou Huang, Huanxiang Liu, Xiaojun Yao
{"title":"深度学习方法在新药物设计和分子动力学模拟中的应用进展","authors":"Qifeng Bai, Shuo Liu, Yanan Tian, Tingyang Xu, Antonio Jesús Banegas-Luna, Horacio Pérez-Sánchez, Junzhou Huang, Huanxiang Liu, Xiaojun Yao","doi":"10.1002/wcms.1581","DOIUrl":null,"url":null,"abstract":"<p>De novo drug design is a stationary way to build novel ligands in the confined pocket of receptor by assembling the atoms or fragments, while molecular dynamics (MD) simulation is a dynamical way to study the interaction mechanism between the ligands and receptors based on the molecular force field. De novo drug design and MD simulation are effective tools for novel drug discovery. With the development of technology, deep learning methods, and interpretable machine learning (IML) have emerged in the research area of drug design. Deep learning methods and IML can be used further to improve the efficiency and accuracy of de novo drug design and MD simulations. The application summary of deep learning methods for de novo drug design, MD simulations, and IML can further promote the technical development of drug discovery. In this article, two major workflow methods and the related components of classical algorithm and deep learning are described for de novo drug design from a new perspective. The application progress of deep learning is also summarized for MD simulations. Furthermore, IML is introduced for the deep learning model interpretability of de novo drug design and MD simulations. Our paper deals with an interesting topic about deep learning applications of de novo drug design and MD simulations for the scientific community.</p><p>This article is categorized under:\n </p>","PeriodicalId":236,"journal":{"name":"Wiley Interdisciplinary Reviews: Computational Molecular Science","volume":"12 3","pages":""},"PeriodicalIF":16.8000,"publicationDate":"2021-10-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/wcms.1581","citationCount":"36","resultStr":"{\"title\":\"Application advances of deep learning methods for de novo drug design and molecular dynamics simulation\",\"authors\":\"Qifeng Bai, Shuo Liu, Yanan Tian, Tingyang Xu, Antonio Jesús Banegas-Luna, Horacio Pérez-Sánchez, Junzhou Huang, Huanxiang Liu, Xiaojun Yao\",\"doi\":\"10.1002/wcms.1581\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>De novo drug design is a stationary way to build novel ligands in the confined pocket of receptor by assembling the atoms or fragments, while molecular dynamics (MD) simulation is a dynamical way to study the interaction mechanism between the ligands and receptors based on the molecular force field. De novo drug design and MD simulation are effective tools for novel drug discovery. With the development of technology, deep learning methods, and interpretable machine learning (IML) have emerged in the research area of drug design. Deep learning methods and IML can be used further to improve the efficiency and accuracy of de novo drug design and MD simulations. The application summary of deep learning methods for de novo drug design, MD simulations, and IML can further promote the technical development of drug discovery. In this article, two major workflow methods and the related components of classical algorithm and deep learning are described for de novo drug design from a new perspective. The application progress of deep learning is also summarized for MD simulations. Furthermore, IML is introduced for the deep learning model interpretability of de novo drug design and MD simulations. Our paper deals with an interesting topic about deep learning applications of de novo drug design and MD simulations for the scientific community.</p><p>This article is categorized under:\\n </p>\",\"PeriodicalId\":236,\"journal\":{\"name\":\"Wiley Interdisciplinary Reviews: Computational Molecular Science\",\"volume\":\"12 3\",\"pages\":\"\"},\"PeriodicalIF\":16.8000,\"publicationDate\":\"2021-10-14\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/wcms.1581\",\"citationCount\":\"36\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Wiley Interdisciplinary Reviews: Computational Molecular Science\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/wcms.1581\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Wiley Interdisciplinary Reviews: Computational Molecular Science","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/wcms.1581","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
Application advances of deep learning methods for de novo drug design and molecular dynamics simulation
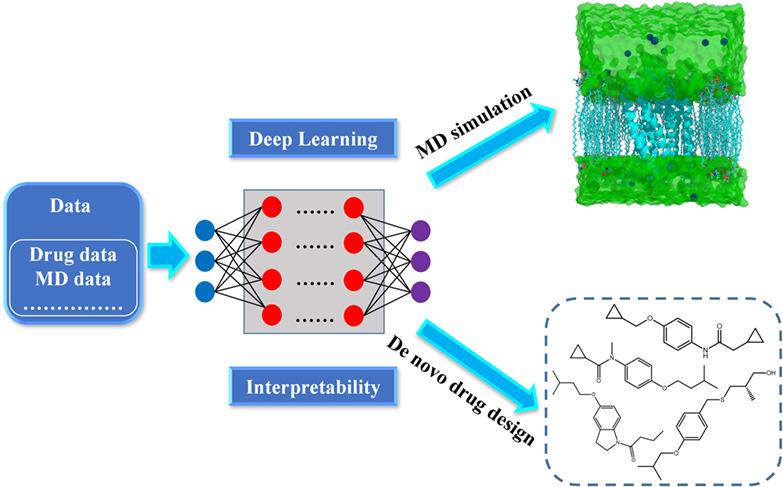
De novo drug design is a stationary way to build novel ligands in the confined pocket of receptor by assembling the atoms or fragments, while molecular dynamics (MD) simulation is a dynamical way to study the interaction mechanism between the ligands and receptors based on the molecular force field. De novo drug design and MD simulation are effective tools for novel drug discovery. With the development of technology, deep learning methods, and interpretable machine learning (IML) have emerged in the research area of drug design. Deep learning methods and IML can be used further to improve the efficiency and accuracy of de novo drug design and MD simulations. The application summary of deep learning methods for de novo drug design, MD simulations, and IML can further promote the technical development of drug discovery. In this article, two major workflow methods and the related components of classical algorithm and deep learning are described for de novo drug design from a new perspective. The application progress of deep learning is also summarized for MD simulations. Furthermore, IML is introduced for the deep learning model interpretability of de novo drug design and MD simulations. Our paper deals with an interesting topic about deep learning applications of de novo drug design and MD simulations for the scientific community.
期刊介绍:
Computational molecular sciences harness the power of rigorous chemical and physical theories, employing computer-based modeling, specialized hardware, software development, algorithm design, and database management to explore and illuminate every facet of molecular sciences. These interdisciplinary approaches form a bridge between chemistry, biology, and materials sciences, establishing connections with adjacent application-driven fields in both chemistry and biology. WIREs Computational Molecular Science stands as a platform to comprehensively review and spotlight research from these dynamic and interconnected fields.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


