Maria E. Yakovleva , Charlotte Welinder , Yutaka Sugihara , Krzysztof Pawłowski , Melinda Rezeli , Elisabet Wieslander , Johan Malm , György Marko-Varga
{"title":"大规模分析黑色素瘤组织样本的工作流程","authors":"Maria E. Yakovleva , Charlotte Welinder , Yutaka Sugihara , Krzysztof Pawłowski , Melinda Rezeli , Elisabet Wieslander , Johan Malm , György Marko-Varga","doi":"10.1016/j.euprot.2015.07.011","DOIUrl":null,"url":null,"abstract":"<div><p>The aim of the present study was to create an optimal workflow for analysing a large cohort of malignant melanoma tissue samples. Samples were lysed with urea and enzymatically digested with trypsin or trypsin/Lys C. Buffer exchange or dilution was used to reduce urea concentration prior to digestion. The tissue digests were analysed directly or following strong cation exchange (SCX) fractionation by nano LC–MS/MS. The approach which resulted in the largest number of protein IDs involved a buffer exchange step before enzymatic digestion with trypsin and chromatographic separation in 120<!--> <!-->min gradient followed by SCX–RP separation of peptides.</p></div>","PeriodicalId":38260,"journal":{"name":"EuPA Open Proteomics","volume":"8 ","pages":"Pages 78-84"},"PeriodicalIF":0.0000,"publicationDate":"2015-09-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1016/j.euprot.2015.07.011","citationCount":"6","resultStr":"{\"title\":\"Workflow for large-scale analysis of melanoma tissue samples\",\"authors\":\"Maria E. Yakovleva , Charlotte Welinder , Yutaka Sugihara , Krzysztof Pawłowski , Melinda Rezeli , Elisabet Wieslander , Johan Malm , György Marko-Varga\",\"doi\":\"10.1016/j.euprot.2015.07.011\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>The aim of the present study was to create an optimal workflow for analysing a large cohort of malignant melanoma tissue samples. Samples were lysed with urea and enzymatically digested with trypsin or trypsin/Lys C. Buffer exchange or dilution was used to reduce urea concentration prior to digestion. The tissue digests were analysed directly or following strong cation exchange (SCX) fractionation by nano LC–MS/MS. The approach which resulted in the largest number of protein IDs involved a buffer exchange step before enzymatic digestion with trypsin and chromatographic separation in 120<!--> <!-->min gradient followed by SCX–RP separation of peptides.</p></div>\",\"PeriodicalId\":38260,\"journal\":{\"name\":\"EuPA Open Proteomics\",\"volume\":\"8 \",\"pages\":\"Pages 78-84\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2015-09-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1016/j.euprot.2015.07.011\",\"citationCount\":\"6\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"EuPA Open Proteomics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S221296851530012X\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"Biochemistry, Genetics and Molecular Biology\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"EuPA Open Proteomics","FirstCategoryId":"1085","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S221296851530012X","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
Workflow for large-scale analysis of melanoma tissue samples
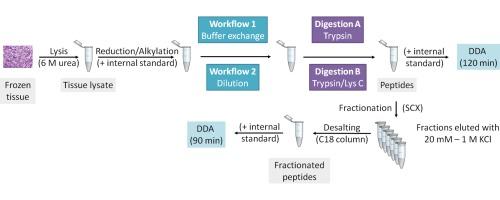
The aim of the present study was to create an optimal workflow for analysing a large cohort of malignant melanoma tissue samples. Samples were lysed with urea and enzymatically digested with trypsin or trypsin/Lys C. Buffer exchange or dilution was used to reduce urea concentration prior to digestion. The tissue digests were analysed directly or following strong cation exchange (SCX) fractionation by nano LC–MS/MS. The approach which resulted in the largest number of protein IDs involved a buffer exchange step before enzymatic digestion with trypsin and chromatographic separation in 120 min gradient followed by SCX–RP separation of peptides.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


