GUIDEMOL:基于RDKit的分子描述符的Python图形用户界面。
IF 3.1
4区 医学
Q3 CHEMISTRY, MEDICINAL
引用次数: 0
摘要
tkinter GUIDEMOL是一个基于RDKit软件的Python计算机程序,用于处理分子结构并使用该包通过图形用户界面计算分子描述符。它可以计算RDKit中已经实现的描述符,以及使用静电势或体素的3D分子结构的网格表示。GUIDEMOL应用程序为没有编程技能的化学信息学用户提供了对RDKit工具的轻松访问,并且可以用于计算其他描述符或触发其他程序。还提供了用于计算网格表示的命令行界面(CLI)。源代码位于https://github.com/jairesdesousa/guidemol.本文章由计算机程序翻译,如有差异,请以英文原文为准。
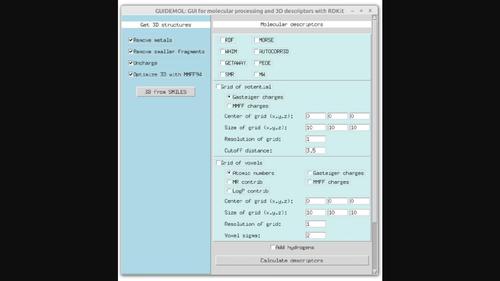
GUIDEMOL: A Python graphical user interface for molecular descriptors based on RDKit.
GUIDEMOL is a Python computer program based on the RDKit software to process molecular structures and calculate molecular descriptors with a graphical user interface using the tkinter package. It can calculate descriptors already implemented in RDKit as well as grid representations of 3D molecular structures using the electrostatic potential or voxels. The GUIDEMOL app provides easy access to RDKit tools for chemoinformatics users with no programming skills and can be adapted to calculate other descriptors or to trigger other procedures. A command line interface (CLI) is also provided for the calculation of grid representations. The source code is available at https://github.com/jairesdesousa/guidemol.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Molecular Informatics
CHEMISTRY, MEDICINAL-MATHEMATICAL & COMPUTATIONAL BIOLOGY
CiteScore
7.30
自引率
2.80%
发文量
70
审稿时长
3 months
期刊介绍:
Molecular Informatics is a peer-reviewed, international forum for publication of high-quality, interdisciplinary research on all molecular aspects of bio/cheminformatics and computer-assisted molecular design. Molecular Informatics succeeded QSAR & Combinatorial Science in 2010.
Molecular Informatics presents methodological innovations that will lead to a deeper understanding of ligand-receptor interactions, macromolecular complexes, molecular networks, design concepts and processes that demonstrate how ideas and design concepts lead to molecules with a desired structure or function, preferably including experimental validation.
The journal''s scope includes but is not limited to the fields of drug discovery and chemical biology, protein and nucleic acid engineering and design, the design of nanomolecular structures, strategies for modeling of macromolecular assemblies, molecular networks and systems, pharmaco- and chemogenomics, computer-assisted screening strategies, as well as novel technologies for the de novo design of biologically active molecules. As a unique feature Molecular Informatics publishes so-called "Methods Corner" review-type articles which feature important technological concepts and advances within the scope of the journal.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


