{"title":"DeepRaccess:使用深度学习进行高速RNA可及性预测。","authors":"Kaisei Hara, Natsuki Iwano, Tsukasa Fukunaga, Michiaki Hamada","doi":"10.3389/fbinf.2023.1275787","DOIUrl":null,"url":null,"abstract":"<p><p>RNA accessibility is a useful RNA secondary structural feature for predicting RNA-RNA interactions and translation efficiency in prokaryotes. However, conventional accessibility calculation tools, such as Raccess, are computationally expensive and require considerable computational time to perform transcriptome-scale analysis. In this study, we developed DeepRaccess, which predicts RNA accessibility based on deep learning methods. DeepRaccess was trained to take artificial RNA sequences as input and to predict the accessibility of these sequences as calculated by Raccess. Simulation and empirical dataset analyses showed that the accessibility predicted by DeepRaccess was highly correlated with the accessibility calculated by Raccess. In addition, we confirmed that DeepRaccess could predict protein abundance in <i>E.coli</i> with moderate accuracy from the sequences around the start codon. We also demonstrated that DeepRaccess achieved tens to hundreds of times software speed-up in a GPU environment. The source codes and the trained models of DeepRaccess are freely available at https://github.com/hmdlab/DeepRaccess.</p>","PeriodicalId":73066,"journal":{"name":"Frontiers in bioinformatics","volume":"3 ","pages":"1275787"},"PeriodicalIF":2.8000,"publicationDate":"2023-10-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10597636/pdf/","citationCount":"0","resultStr":"{\"title\":\"DeepRaccess: high-speed RNA accessibility prediction using deep learning.\",\"authors\":\"Kaisei Hara, Natsuki Iwano, Tsukasa Fukunaga, Michiaki Hamada\",\"doi\":\"10.3389/fbinf.2023.1275787\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>RNA accessibility is a useful RNA secondary structural feature for predicting RNA-RNA interactions and translation efficiency in prokaryotes. However, conventional accessibility calculation tools, such as Raccess, are computationally expensive and require considerable computational time to perform transcriptome-scale analysis. In this study, we developed DeepRaccess, which predicts RNA accessibility based on deep learning methods. DeepRaccess was trained to take artificial RNA sequences as input and to predict the accessibility of these sequences as calculated by Raccess. Simulation and empirical dataset analyses showed that the accessibility predicted by DeepRaccess was highly correlated with the accessibility calculated by Raccess. In addition, we confirmed that DeepRaccess could predict protein abundance in <i>E.coli</i> with moderate accuracy from the sequences around the start codon. We also demonstrated that DeepRaccess achieved tens to hundreds of times software speed-up in a GPU environment. The source codes and the trained models of DeepRaccess are freely available at https://github.com/hmdlab/DeepRaccess.</p>\",\"PeriodicalId\":73066,\"journal\":{\"name\":\"Frontiers in bioinformatics\",\"volume\":\"3 \",\"pages\":\"1275787\"},\"PeriodicalIF\":2.8000,\"publicationDate\":\"2023-10-10\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10597636/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Frontiers in bioinformatics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.3389/fbinf.2023.1275787\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q2\",\"JCRName\":\"MATHEMATICAL & COMPUTATIONAL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Frontiers in bioinformatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3389/fbinf.2023.1275787","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
DeepRaccess: high-speed RNA accessibility prediction using deep learning.
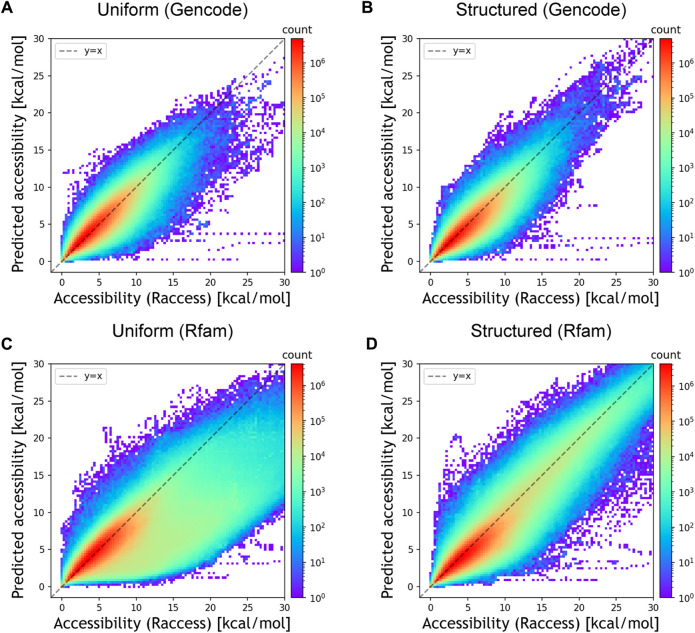
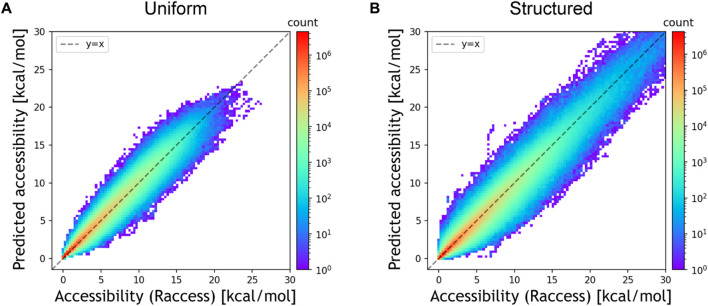
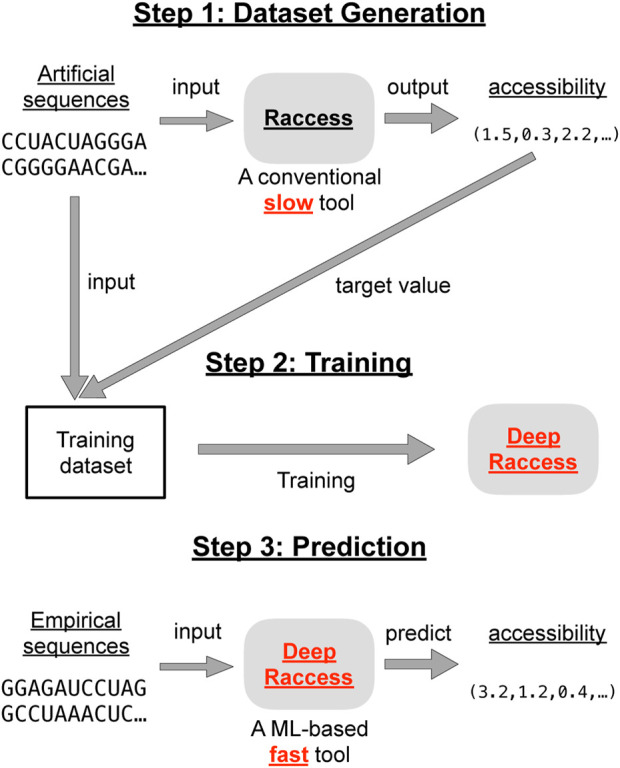
RNA accessibility is a useful RNA secondary structural feature for predicting RNA-RNA interactions and translation efficiency in prokaryotes. However, conventional accessibility calculation tools, such as Raccess, are computationally expensive and require considerable computational time to perform transcriptome-scale analysis. In this study, we developed DeepRaccess, which predicts RNA accessibility based on deep learning methods. DeepRaccess was trained to take artificial RNA sequences as input and to predict the accessibility of these sequences as calculated by Raccess. Simulation and empirical dataset analyses showed that the accessibility predicted by DeepRaccess was highly correlated with the accessibility calculated by Raccess. In addition, we confirmed that DeepRaccess could predict protein abundance in E.coli with moderate accuracy from the sequences around the start codon. We also demonstrated that DeepRaccess achieved tens to hundreds of times software speed-up in a GPU environment. The source codes and the trained models of DeepRaccess are freely available at https://github.com/hmdlab/DeepRaccess.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


