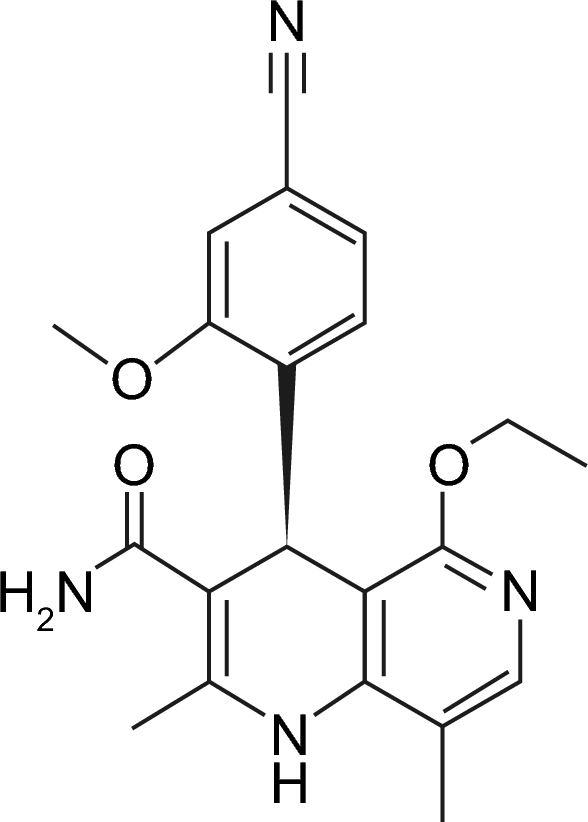
{"title":"非甾体矿物皮质激素受体拮抗剂芬纳酮的药代动力学。","authors":"Roland Heinig, Thomas Eissing","doi":"10.1007/s40262-023-01312-9","DOIUrl":null,"url":null,"abstract":"<p><p>Finerenone, a selective and nonsteroidal antagonist of the mineralocorticoid receptor, has received regulatory approval with the indication of cardiorenal protection in patients with chronic kidney disease associated with type 2 diabetes. It is rapidly and completely absorbed and undergoes first-pass metabolism in the gut wall and liver resulting in a bioavailability of 43.5%. Finerenone can be taken with or without food. The pharmacokinetics of finerenone are linear and its half-life is 2 to 3 h in the dose range of up to 20 mg. Cytochrome P450 (CYP) 3A4 (90%) and CYP2C8 (10%) are involved in the extensive biotransformation of finerenone to pharmacologically inactive metabolites, which are excreted via both renal (80%) and biliary (20%) routes. Moderate or severe renal impairment, or moderate hepatic impairment result in area-under-the-curve increases of finerenone (< 40%), which do not require a dose adjustment per se, as the starting dose is based on estimated glomerular filtration rate (eGFR) and titrated according to serum potassium levels and eGFR decline. No relevant effects of age, sex, body size or ethnicity on systemic finerenone exposure were identified. Modulators of CYP3A4 activity were found to affect finerenone exposure, consistent with its classification as a sensitive CYP3A4 substrate. Serum potassium should be monitored during drug initiation or dosage adjustment of either a moderate or weak CYP3A4 inhibitor or finerenone, and the dose of finerenone should be adjusted as appropriate. Its use with strong inhibitors is contraindicated and strong or moderate inducers of CYP3A4 should be avoided. Finerenone has no potential to affect relevant CYP enzymes and drug transporters.</p>","PeriodicalId":10405,"journal":{"name":"Clinical Pharmacokinetics","volume":null,"pages":null},"PeriodicalIF":4.6000,"publicationDate":"2023-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10684710/pdf/","citationCount":"0","resultStr":"{\"title\":\"The Pharmacokinetics of the Nonsteroidal Mineralocorticoid Receptor Antagonist Finerenone.\",\"authors\":\"Roland Heinig, Thomas Eissing\",\"doi\":\"10.1007/s40262-023-01312-9\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Finerenone, a selective and nonsteroidal antagonist of the mineralocorticoid receptor, has received regulatory approval with the indication of cardiorenal protection in patients with chronic kidney disease associated with type 2 diabetes. It is rapidly and completely absorbed and undergoes first-pass metabolism in the gut wall and liver resulting in a bioavailability of 43.5%. Finerenone can be taken with or without food. The pharmacokinetics of finerenone are linear and its half-life is 2 to 3 h in the dose range of up to 20 mg. Cytochrome P450 (CYP) 3A4 (90%) and CYP2C8 (10%) are involved in the extensive biotransformation of finerenone to pharmacologically inactive metabolites, which are excreted via both renal (80%) and biliary (20%) routes. Moderate or severe renal impairment, or moderate hepatic impairment result in area-under-the-curve increases of finerenone (< 40%), which do not require a dose adjustment per se, as the starting dose is based on estimated glomerular filtration rate (eGFR) and titrated according to serum potassium levels and eGFR decline. No relevant effects of age, sex, body size or ethnicity on systemic finerenone exposure were identified. Modulators of CYP3A4 activity were found to affect finerenone exposure, consistent with its classification as a sensitive CYP3A4 substrate. Serum potassium should be monitored during drug initiation or dosage adjustment of either a moderate or weak CYP3A4 inhibitor or finerenone, and the dose of finerenone should be adjusted as appropriate. Its use with strong inhibitors is contraindicated and strong or moderate inducers of CYP3A4 should be avoided. Finerenone has no potential to affect relevant CYP enzymes and drug transporters.</p>\",\"PeriodicalId\":10405,\"journal\":{\"name\":\"Clinical Pharmacokinetics\",\"volume\":null,\"pages\":null},\"PeriodicalIF\":4.6000,\"publicationDate\":\"2023-12-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10684710/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Clinical Pharmacokinetics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1007/s40262-023-01312-9\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/10/24 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q1\",\"JCRName\":\"PHARMACOLOGY & PHARMACY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical Pharmacokinetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s40262-023-01312-9","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/10/24 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"PHARMACOLOGY & PHARMACY","Score":null,"Total":0}
The Pharmacokinetics of the Nonsteroidal Mineralocorticoid Receptor Antagonist Finerenone.
Finerenone, a selective and nonsteroidal antagonist of the mineralocorticoid receptor, has received regulatory approval with the indication of cardiorenal protection in patients with chronic kidney disease associated with type 2 diabetes. It is rapidly and completely absorbed and undergoes first-pass metabolism in the gut wall and liver resulting in a bioavailability of 43.5%. Finerenone can be taken with or without food. The pharmacokinetics of finerenone are linear and its half-life is 2 to 3 h in the dose range of up to 20 mg. Cytochrome P450 (CYP) 3A4 (90%) and CYP2C8 (10%) are involved in the extensive biotransformation of finerenone to pharmacologically inactive metabolites, which are excreted via both renal (80%) and biliary (20%) routes. Moderate or severe renal impairment, or moderate hepatic impairment result in area-under-the-curve increases of finerenone (< 40%), which do not require a dose adjustment per se, as the starting dose is based on estimated glomerular filtration rate (eGFR) and titrated according to serum potassium levels and eGFR decline. No relevant effects of age, sex, body size or ethnicity on systemic finerenone exposure were identified. Modulators of CYP3A4 activity were found to affect finerenone exposure, consistent with its classification as a sensitive CYP3A4 substrate. Serum potassium should be monitored during drug initiation or dosage adjustment of either a moderate or weak CYP3A4 inhibitor or finerenone, and the dose of finerenone should be adjusted as appropriate. Its use with strong inhibitors is contraindicated and strong or moderate inducers of CYP3A4 should be avoided. Finerenone has no potential to affect relevant CYP enzymes and drug transporters.
期刊介绍:
Clinical Pharmacokinetics promotes the continuing development of clinical pharmacokinetics and pharmacodynamics for the improvement of drug therapy, and for furthering postgraduate education in clinical pharmacology and therapeutics.
Pharmacokinetics, the study of drug disposition in the body, is an integral part of drug development and rational use. Knowledge and application of pharmacokinetic principles leads to accelerated drug development, cost effective drug use and a reduced frequency of adverse effects and drug interactions.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


