{"title":"吡哆醇依赖性癫痫的研究现状与问题","authors":"Curtis R. Coughlin II, Sidney M. Gospe Jr.","doi":"10.1002/cns3.20016","DOIUrl":null,"url":null,"abstract":"Pyridoxine‐dependent epilepsy (PDE) was historically defined by a dramatic clinical response to a trial of pyridoxine and the re‐emergence of seizures after withdrawal of pyridoxine. Research conducted over the last seven decades has revealed that the phenotype of PDE results from multiple genetic disorders, and the most common disorder, PDE‐ALDH7A1, is caused by a deficiency of an enzyme involved in lysine metabolism. PDE‐ALDH7A1 is characterized by more than epilepsy, as many patients have abnormalities of brain development, and most patients have intellectual and developmental disability. Treatment aimed at the underlying metabolic defect, in addition to pyridoxine supplementation, has improved clinical outcomes. Recently discovered biomarkers and genetic testing allow for the diagnosis of PDE‐ALDH7A1 without the need of a pyridoxine trial and hold the promise for newborn screening. Despite these many advances, PDE‐ALDH7A1 remains a clinical and biochemical conundrum. The increasing use of model systems and an international collaboration of clinician‐scientists are among the reasons to be optimistic that these questions will be answered in the near future and that the clinical outcomes and quality of life will continue to improve for patients with PDE‐ALDH7A1.","PeriodicalId":72232,"journal":{"name":"Annals of the Child Neurology Society","volume":"1 1","pages":"24-37"},"PeriodicalIF":0.0000,"publicationDate":"2023-03-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/cns3.20016","citationCount":"1","resultStr":"{\"title\":\"Pyridoxine-dependent epilepsy: Current perspectives and questions for future research\",\"authors\":\"Curtis R. Coughlin II, Sidney M. Gospe Jr.\",\"doi\":\"10.1002/cns3.20016\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"Pyridoxine‐dependent epilepsy (PDE) was historically defined by a dramatic clinical response to a trial of pyridoxine and the re‐emergence of seizures after withdrawal of pyridoxine. Research conducted over the last seven decades has revealed that the phenotype of PDE results from multiple genetic disorders, and the most common disorder, PDE‐ALDH7A1, is caused by a deficiency of an enzyme involved in lysine metabolism. PDE‐ALDH7A1 is characterized by more than epilepsy, as many patients have abnormalities of brain development, and most patients have intellectual and developmental disability. Treatment aimed at the underlying metabolic defect, in addition to pyridoxine supplementation, has improved clinical outcomes. Recently discovered biomarkers and genetic testing allow for the diagnosis of PDE‐ALDH7A1 without the need of a pyridoxine trial and hold the promise for newborn screening. Despite these many advances, PDE‐ALDH7A1 remains a clinical and biochemical conundrum. The increasing use of model systems and an international collaboration of clinician‐scientists are among the reasons to be optimistic that these questions will be answered in the near future and that the clinical outcomes and quality of life will continue to improve for patients with PDE‐ALDH7A1.\",\"PeriodicalId\":72232,\"journal\":{\"name\":\"Annals of the Child Neurology Society\",\"volume\":\"1 1\",\"pages\":\"24-37\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2023-03-07\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/cns3.20016\",\"citationCount\":\"1\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Annals of the Child Neurology Society\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/cns3.20016\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Annals of the Child Neurology Society","FirstCategoryId":"1085","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/cns3.20016","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
Pyridoxine-dependent epilepsy: Current perspectives and questions for future research
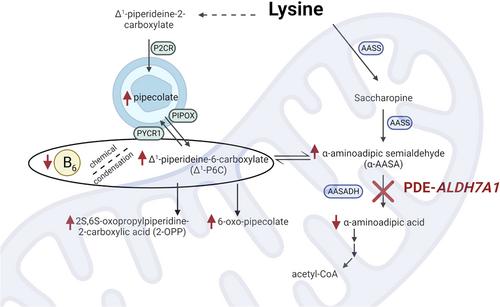
Pyridoxine‐dependent epilepsy (PDE) was historically defined by a dramatic clinical response to a trial of pyridoxine and the re‐emergence of seizures after withdrawal of pyridoxine. Research conducted over the last seven decades has revealed that the phenotype of PDE results from multiple genetic disorders, and the most common disorder, PDE‐ALDH7A1, is caused by a deficiency of an enzyme involved in lysine metabolism. PDE‐ALDH7A1 is characterized by more than epilepsy, as many patients have abnormalities of brain development, and most patients have intellectual and developmental disability. Treatment aimed at the underlying metabolic defect, in addition to pyridoxine supplementation, has improved clinical outcomes. Recently discovered biomarkers and genetic testing allow for the diagnosis of PDE‐ALDH7A1 without the need of a pyridoxine trial and hold the promise for newborn screening. Despite these many advances, PDE‐ALDH7A1 remains a clinical and biochemical conundrum. The increasing use of model systems and an international collaboration of clinician‐scientists are among the reasons to be optimistic that these questions will be answered in the near future and that the clinical outcomes and quality of life will continue to improve for patients with PDE‐ALDH7A1.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


