用于组织蛋白质组图谱分析中细胞类型混合物解卷积的深域对抗神经网络
IF 18.8
1区 计算机科学
Q1 COMPUTER SCIENCE, ARTIFICIAL INTELLIGENCE
引用次数: 0
摘要
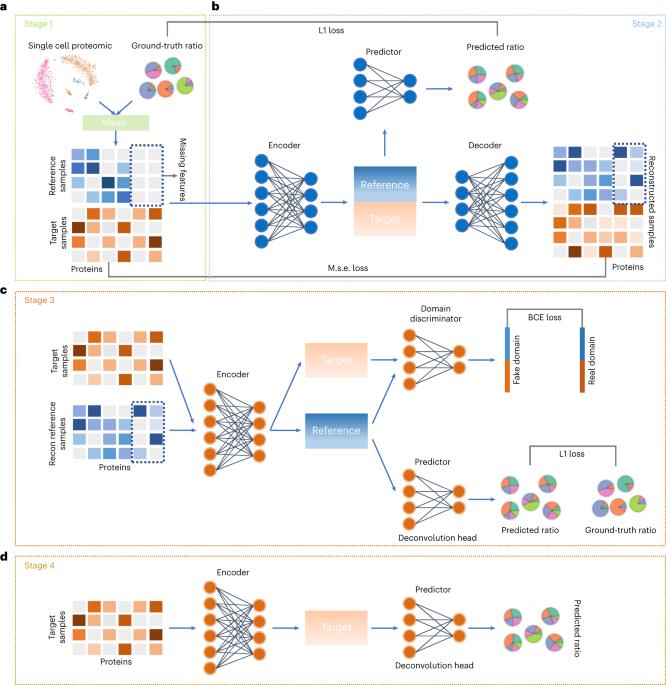
细胞类型解卷积是一种从大量测序数据中确定/分辨细胞类型比例的计算方法,常用于分析肿瘤组织样本中的不同细胞类型。然而,由于可重复性/再现性、可变参考标准和单细胞蛋白质组参考数据的缺乏,去卷积技术在利用蛋白质组数据分析细胞类型方面仍处于起步阶段。scpDeconv 使用自动编码器来利用批量蛋白质组数据的信息来提高单细胞蛋白质组数据的质量,并采用域对抗架构来连接单细胞和批量数据分布,并将标签从单细胞数据转移到批量数据。大量实验验证了 scpDeconv 在对不同物种/来源和不同蛋白质组技术产生的蛋白质组数据进行解卷积时的性能。该方法可广泛应用于肿瘤微环境解读和临床诊断/分类等领域。对生物信息学界来说,对组织蛋白质组数据中的细胞类型进行解卷积是一项具有挑战性的计算任务。本文介绍了一种称为 scpDeconv 的深度学习方法,它能有效利用单细胞蛋白质组学数据,从大量蛋白质组学测量中解构细胞类型和状态。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Deep domain adversarial neural network for the deconvolution of cell type mixtures in tissue proteome profiling
Cell type deconvolution is a computational method for the determination/resolution of cell type proportions from bulk sequencing data, and is frequently used for the analysis of divergent cell types in tumour tissue samples. However, deconvolution technology is still in its infancy for the analysis of cell types using proteomic data due to challenges with repeatability/reproducibility, variable reference standards and the lack of single-cell proteomic reference data. Here we develop a deep-learning-based deconvolution method (scpDeconv) specifically designed for proteomic data. scpDeconv uses an autoencoder to leverage the information from bulk proteomic data to improve the quality of single-cell proteomic data, and employs a domain adversarial architecture to bridge the single-cell and bulk data distributions and transfer labels from single-cell data to bulk data. Extensive experiments validate the performance of scpDeconv in the deconvolution of proteomic data produced from various species/sources and different proteomic technologies. This method should find broad applicability to areas including tumour microenvironment interpretation and clinical diagnosis/classification. Deconvolution of cell types in tissue proteomic data is a challenging computational task for the bioinformatics community. A deep-learning method termed scpDeconv is introduced that makes efficient use of single-cell proteomics data to deconvolve cell types and states from bulk proteomics measurements.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Nature Machine Intelligence
Multiple-
CiteScore
36.90
自引率
2.10%
发文量
127
期刊介绍:
Nature Machine Intelligence is a distinguished publication that presents original research and reviews on various topics in machine learning, robotics, and AI. Our focus extends beyond these fields, exploring their profound impact on other scientific disciplines, as well as societal and industrial aspects. We recognize limitless possibilities wherein machine intelligence can augment human capabilities and knowledge in domains like scientific exploration, healthcare, medical diagnostics, and the creation of safe and sustainable cities, transportation, and agriculture. Simultaneously, we acknowledge the emergence of ethical, social, and legal concerns due to the rapid pace of advancements.
To foster interdisciplinary discussions on these far-reaching implications, Nature Machine Intelligence serves as a platform for dialogue facilitated through Comments, News Features, News & Views articles, and Correspondence. Our goal is to encourage a comprehensive examination of these subjects.
Similar to all Nature-branded journals, Nature Machine Intelligence operates under the guidance of a team of skilled editors. We adhere to a fair and rigorous peer-review process, ensuring high standards of copy-editing and production, swift publication, and editorial independence.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


