Christophe Ambroise, Alia Dehman, Pierre Neuvial, Guillem Rigaill, Nathalie Vialaneix
{"title":"带相似性矩阵的邻接约束层次聚类及其在基因组学中的应用。","authors":"Christophe Ambroise, Alia Dehman, Pierre Neuvial, Guillem Rigaill, Nathalie Vialaneix","doi":"10.1186/s13015-019-0157-4","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Genomic data analyses such as Genome-Wide Association Studies (GWAS) or Hi-C studies are often faced with the problem of partitioning chromosomes into successive regions based on a similarity matrix of high-resolution, locus-level measurements. An intuitive way of doing this is to perform a modified Hierarchical Agglomerative Clustering (HAC), where only adjacent clusters (according to the ordering of positions within a chromosome) are allowed to be merged. But a major practical drawback of this method is its quadratic time and space complexity in the number of loci, which is typically of the order of <math><msup><mn>10</mn> <mn>4</mn></msup> </math> to <math><msup><mn>10</mn> <mn>5</mn></msup> </math> for each chromosome.</p><p><strong>Results: </strong>By assuming that the similarity between physically distant objects is negligible, we are able to propose an implementation of adjacency-constrained HAC with quasi-linear complexity. This is achieved by pre-calculating specific sums of similarities, and storing candidate fusions in a min-heap. Our illustrations on GWAS and Hi-C datasets demonstrate the relevance of this assumption, and show that this method highlights biologically meaningful signals. Thanks to its small time and memory footprint, the method can be run on a standard laptop in minutes or even seconds.</p><p><strong>Availability and implementation: </strong>Software and sample data are available as an R package, <b>adjclust</b>, that can be downloaded from the Comprehensive R Archive Network (CRAN).</p>","PeriodicalId":50823,"journal":{"name":"Algorithms for Molecular Biology","volume":"14 ","pages":"22"},"PeriodicalIF":1.7000,"publicationDate":"2019-11-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s13015-019-0157-4","citationCount":"21","resultStr":"{\"title\":\"Adjacency-constrained hierarchical clustering of a band similarity matrix with application to genomics.\",\"authors\":\"Christophe Ambroise, Alia Dehman, Pierre Neuvial, Guillem Rigaill, Nathalie Vialaneix\",\"doi\":\"10.1186/s13015-019-0157-4\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Genomic data analyses such as Genome-Wide Association Studies (GWAS) or Hi-C studies are often faced with the problem of partitioning chromosomes into successive regions based on a similarity matrix of high-resolution, locus-level measurements. An intuitive way of doing this is to perform a modified Hierarchical Agglomerative Clustering (HAC), where only adjacent clusters (according to the ordering of positions within a chromosome) are allowed to be merged. But a major practical drawback of this method is its quadratic time and space complexity in the number of loci, which is typically of the order of <math><msup><mn>10</mn> <mn>4</mn></msup> </math> to <math><msup><mn>10</mn> <mn>5</mn></msup> </math> for each chromosome.</p><p><strong>Results: </strong>By assuming that the similarity between physically distant objects is negligible, we are able to propose an implementation of adjacency-constrained HAC with quasi-linear complexity. This is achieved by pre-calculating specific sums of similarities, and storing candidate fusions in a min-heap. Our illustrations on GWAS and Hi-C datasets demonstrate the relevance of this assumption, and show that this method highlights biologically meaningful signals. Thanks to its small time and memory footprint, the method can be run on a standard laptop in minutes or even seconds.</p><p><strong>Availability and implementation: </strong>Software and sample data are available as an R package, <b>adjclust</b>, that can be downloaded from the Comprehensive R Archive Network (CRAN).</p>\",\"PeriodicalId\":50823,\"journal\":{\"name\":\"Algorithms for Molecular Biology\",\"volume\":\"14 \",\"pages\":\"22\"},\"PeriodicalIF\":1.7000,\"publicationDate\":\"2019-11-15\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/s13015-019-0157-4\",\"citationCount\":\"21\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Algorithms for Molecular Biology\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1186/s13015-019-0157-4\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2019/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q4\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Algorithms for Molecular Biology","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s13015-019-0157-4","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2019/1/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
Adjacency-constrained hierarchical clustering of a band similarity matrix with application to genomics.
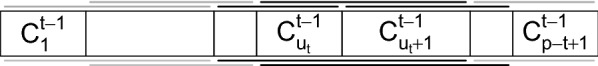
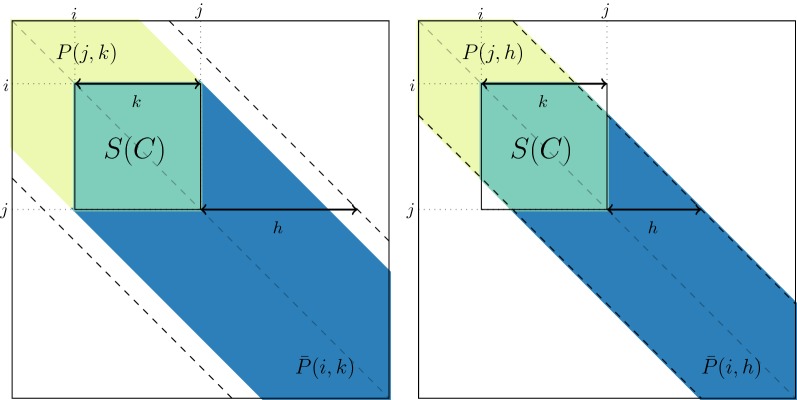
Background: Genomic data analyses such as Genome-Wide Association Studies (GWAS) or Hi-C studies are often faced with the problem of partitioning chromosomes into successive regions based on a similarity matrix of high-resolution, locus-level measurements. An intuitive way of doing this is to perform a modified Hierarchical Agglomerative Clustering (HAC), where only adjacent clusters (according to the ordering of positions within a chromosome) are allowed to be merged. But a major practical drawback of this method is its quadratic time and space complexity in the number of loci, which is typically of the order of to for each chromosome.
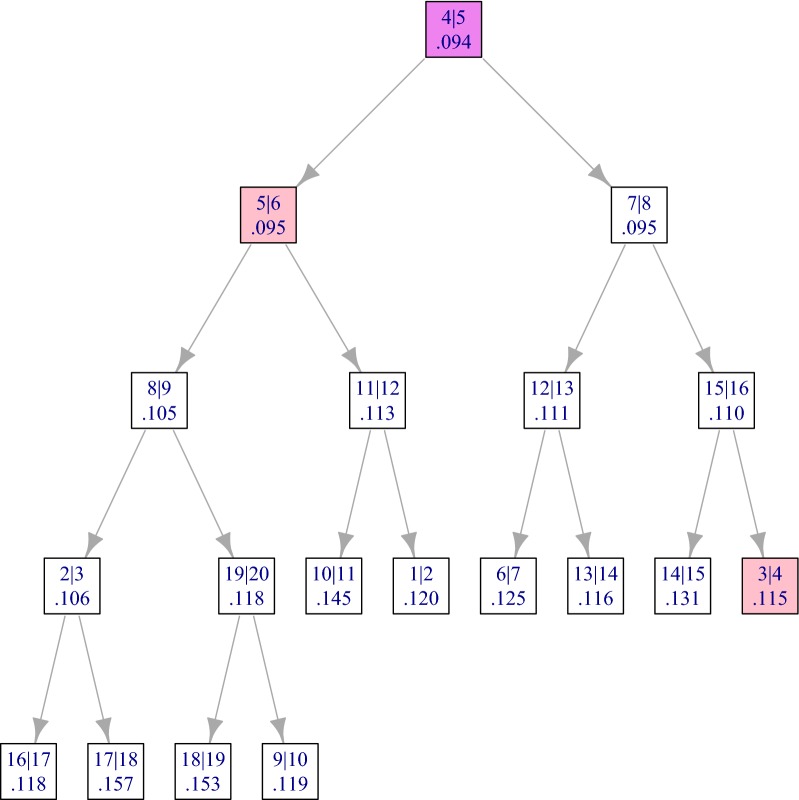
Results: By assuming that the similarity between physically distant objects is negligible, we are able to propose an implementation of adjacency-constrained HAC with quasi-linear complexity. This is achieved by pre-calculating specific sums of similarities, and storing candidate fusions in a min-heap. Our illustrations on GWAS and Hi-C datasets demonstrate the relevance of this assumption, and show that this method highlights biologically meaningful signals. Thanks to its small time and memory footprint, the method can be run on a standard laptop in minutes or even seconds.
Availability and implementation: Software and sample data are available as an R package, adjclust, that can be downloaded from the Comprehensive R Archive Network (CRAN).
期刊介绍:
Algorithms for Molecular Biology publishes articles on novel algorithms for biological sequence and structure analysis, phylogeny reconstruction, and combinatorial algorithms and machine learning.
Areas of interest include but are not limited to: algorithms for RNA and protein structure analysis, gene prediction and genome analysis, comparative sequence analysis and alignment, phylogeny, gene expression, machine learning, and combinatorial algorithms.
Where appropriate, manuscripts should describe applications to real-world data. However, pure algorithm papers are also welcome if future applications to biological data are to be expected, or if they address complexity or approximation issues of novel computational problems in molecular biology. Articles about novel software tools will be considered for publication if they contain some algorithmically interesting aspects.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


