{"title":"一种新的PET配体GSK’963类似物的合成和评估,旨在脑中受体相互作用蛋白激酶1的放射自显影和成像。","authors":"Hiroshi Ikenuma, Aya Ogata, Hiroko Koyama, Bin Ji, Hideki Ishii, Takashi Yamada, Junichiro Abe, Chie Seki, Yuji Nagai, Masanori Ichise, Takafumi Minamimoto, Makoto Higuchi, Ming-Rong Zhang, Takashi Kato, Kengo Ito, Masaaki Suzuki, Yasuyuki Kimura","doi":"10.1186/s41181-023-00217-z","DOIUrl":null,"url":null,"abstract":"<div><h3>Background</h3><p>Receptor interacting protein kinase 1 (RIPK1) is a serine/threonine kinase, which regulates programmed cell death and inflammation. Recently, the involvement of RIPK1 in the pathophysiology of Alzheimer’s disease (AD) has been reported; RIPK1 is involved in microglia’s phenotypic transition to their dysfunctional states, and it is highly expressed in the neurons and microglia in the postmortem brains in AD patients. They prompt neurodegeneration leading to accumulations of pathological proteins in AD. Therefore, regulation of RIPK1 could be a potential therapeutic target for the treatment of AD, and in vivo imaging of RIPK1 may become a useful modality in studies of drug discovery and pathophysiology of AD. The purpose of this study was to develop a suitable radioligand for positron emission tomography (PET) imaging of RIPK1.</p><h3>Results</h3><p>(<i>S</i>)-2,2-dimethyl-1-(5-phenyl-4,5-dihydro-1<i>H</i>-pyrazol-1-yl)propan-1-one (GSK’963) has a high affinity, selectivity for RIPK1, and favorable physiochemical properties based on its chemical structure. In this study, since <sup>11</sup>C-labeling (half-life: 20.4 min) GSK’963 retaining its structure requiring the Grignard reaction of <i>tert</i>-butylmagnesium halides and [<sup>11</sup>C]carbon dioxide was anticipated to give a low yield, we decided instead to <sup>11</sup>C-label a GSK’963 analog ((<i>S</i>)-2,2-dimethyl-1-(5-(<i>m</i>-tolyl)-4,5-dihydro-1<i>H</i>-pyrazol-1-yl)propan-1-one, GG502), which has a high RIPK1 inhibitory activity equivalent to that of the original compound GSK’963. Thus, we successfully <sup>11</sup>C-labeled GG502 using a Pd-mediated cross-coupling reaction in favorable yields (3.6 ± 1.9%) and radiochemical purities (> 96%), and molar activity (47–115 GBq/μmol). On autoradiography, radioactivity accumulation was observed for [<sup>11</sup>C]GG502 and decreased by non-radioactive GG502 in the mouse spleen and human brain, indicating the possibility of specific binding of this ligand to RIPK1. On brain PET imaging in a rhesus monkey, [<sup>11</sup>C]GG502 showed a good brain permeability (peak standardized uptake value (SUV) ~3.0), although there was no clear evidence of specific binding of [<sup>11</sup>C]GG502. On brain PET imaging in acute inflammation model rats, [<sup>11</sup>C]GG502 also showed a good brain permeability, and no significant increased uptake was observed in the lipopolysaccharide-treated side of striatum. On metabolite analysis in rats at 30 min after administration of [<sup>11</sup>C]GG502, ~55% and ~10% of radioactivity was from unmetabolized [<sup>11</sup>C]GG502 in the brain and the plasma, respectively.</p><h3>Conclusions</h3><p>We synthesized and evaluated a <sup>11</sup>C-labeled PET ligand based on the methylated analog of GSK’963 for imaging of RIPK1 in the brain. Although in autoradiography of the resulting [<sup>11</sup>C]GG502 indicated the possibility of specific binding, the actual PET imaging failed to detect any evidence of specific binding to RIPK1 despite its good brain permeability. Further development of radioligands with a higher binding affinity for RIPK1 in vivo and more stable metabolite profiles compared with the current compound may be required.</p></div>","PeriodicalId":534,"journal":{"name":"EJNMMI Radiopharmacy and Chemistry","volume":"8 1","pages":""},"PeriodicalIF":4.4000,"publicationDate":"2023-10-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10584749/pdf/","citationCount":"0","resultStr":"{\"title\":\"Synthesis and evaluation of a novel PET ligand, a GSK’963 analog, aiming at autoradiography and imaging of the receptor interacting protein kinase 1 in the brain\",\"authors\":\"Hiroshi Ikenuma, Aya Ogata, Hiroko Koyama, Bin Ji, Hideki Ishii, Takashi Yamada, Junichiro Abe, Chie Seki, Yuji Nagai, Masanori Ichise, Takafumi Minamimoto, Makoto Higuchi, Ming-Rong Zhang, Takashi Kato, Kengo Ito, Masaaki Suzuki, Yasuyuki Kimura\",\"doi\":\"10.1186/s41181-023-00217-z\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><h3>Background</h3><p>Receptor interacting protein kinase 1 (RIPK1) is a serine/threonine kinase, which regulates programmed cell death and inflammation. Recently, the involvement of RIPK1 in the pathophysiology of Alzheimer’s disease (AD) has been reported; RIPK1 is involved in microglia’s phenotypic transition to their dysfunctional states, and it is highly expressed in the neurons and microglia in the postmortem brains in AD patients. They prompt neurodegeneration leading to accumulations of pathological proteins in AD. Therefore, regulation of RIPK1 could be a potential therapeutic target for the treatment of AD, and in vivo imaging of RIPK1 may become a useful modality in studies of drug discovery and pathophysiology of AD. The purpose of this study was to develop a suitable radioligand for positron emission tomography (PET) imaging of RIPK1.</p><h3>Results</h3><p>(<i>S</i>)-2,2-dimethyl-1-(5-phenyl-4,5-dihydro-1<i>H</i>-pyrazol-1-yl)propan-1-one (GSK’963) has a high affinity, selectivity for RIPK1, and favorable physiochemical properties based on its chemical structure. In this study, since <sup>11</sup>C-labeling (half-life: 20.4 min) GSK’963 retaining its structure requiring the Grignard reaction of <i>tert</i>-butylmagnesium halides and [<sup>11</sup>C]carbon dioxide was anticipated to give a low yield, we decided instead to <sup>11</sup>C-label a GSK’963 analog ((<i>S</i>)-2,2-dimethyl-1-(5-(<i>m</i>-tolyl)-4,5-dihydro-1<i>H</i>-pyrazol-1-yl)propan-1-one, GG502), which has a high RIPK1 inhibitory activity equivalent to that of the original compound GSK’963. Thus, we successfully <sup>11</sup>C-labeled GG502 using a Pd-mediated cross-coupling reaction in favorable yields (3.6 ± 1.9%) and radiochemical purities (> 96%), and molar activity (47–115 GBq/μmol). On autoradiography, radioactivity accumulation was observed for [<sup>11</sup>C]GG502 and decreased by non-radioactive GG502 in the mouse spleen and human brain, indicating the possibility of specific binding of this ligand to RIPK1. On brain PET imaging in a rhesus monkey, [<sup>11</sup>C]GG502 showed a good brain permeability (peak standardized uptake value (SUV) ~3.0), although there was no clear evidence of specific binding of [<sup>11</sup>C]GG502. On brain PET imaging in acute inflammation model rats, [<sup>11</sup>C]GG502 also showed a good brain permeability, and no significant increased uptake was observed in the lipopolysaccharide-treated side of striatum. On metabolite analysis in rats at 30 min after administration of [<sup>11</sup>C]GG502, ~55% and ~10% of radioactivity was from unmetabolized [<sup>11</sup>C]GG502 in the brain and the plasma, respectively.</p><h3>Conclusions</h3><p>We synthesized and evaluated a <sup>11</sup>C-labeled PET ligand based on the methylated analog of GSK’963 for imaging of RIPK1 in the brain. Although in autoradiography of the resulting [<sup>11</sup>C]GG502 indicated the possibility of specific binding, the actual PET imaging failed to detect any evidence of specific binding to RIPK1 despite its good brain permeability. Further development of radioligands with a higher binding affinity for RIPK1 in vivo and more stable metabolite profiles compared with the current compound may be required.</p></div>\",\"PeriodicalId\":534,\"journal\":{\"name\":\"EJNMMI Radiopharmacy and Chemistry\",\"volume\":\"8 1\",\"pages\":\"\"},\"PeriodicalIF\":4.4000,\"publicationDate\":\"2023-10-18\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10584749/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"EJNMMI Radiopharmacy and Chemistry\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://link.springer.com/article/10.1186/s41181-023-00217-z\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, INORGANIC & NUCLEAR\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"EJNMMI Radiopharmacy and Chemistry","FirstCategoryId":"1085","ListUrlMain":"https://link.springer.com/article/10.1186/s41181-023-00217-z","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, INORGANIC & NUCLEAR","Score":null,"Total":0}
Synthesis and evaluation of a novel PET ligand, a GSK’963 analog, aiming at autoradiography and imaging of the receptor interacting protein kinase 1 in the brain
Background
Receptor interacting protein kinase 1 (RIPK1) is a serine/threonine kinase, which regulates programmed cell death and inflammation. Recently, the involvement of RIPK1 in the pathophysiology of Alzheimer’s disease (AD) has been reported; RIPK1 is involved in microglia’s phenotypic transition to their dysfunctional states, and it is highly expressed in the neurons and microglia in the postmortem brains in AD patients. They prompt neurodegeneration leading to accumulations of pathological proteins in AD. Therefore, regulation of RIPK1 could be a potential therapeutic target for the treatment of AD, and in vivo imaging of RIPK1 may become a useful modality in studies of drug discovery and pathophysiology of AD. The purpose of this study was to develop a suitable radioligand for positron emission tomography (PET) imaging of RIPK1.
Results
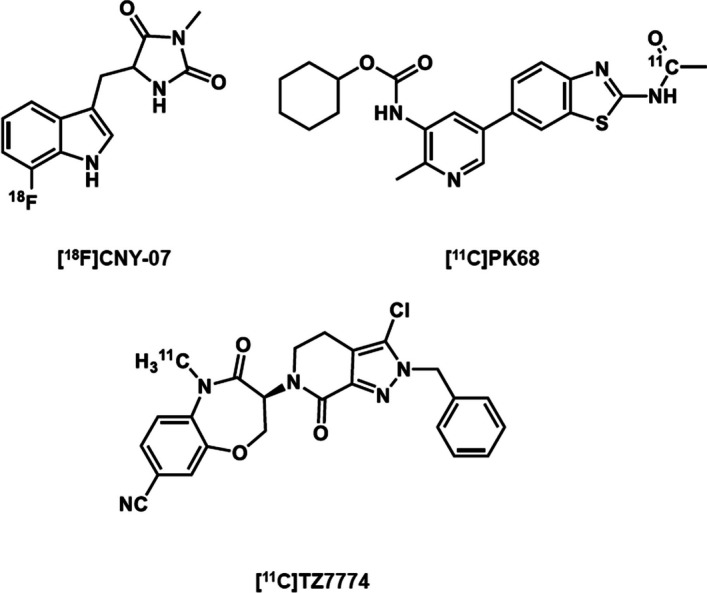
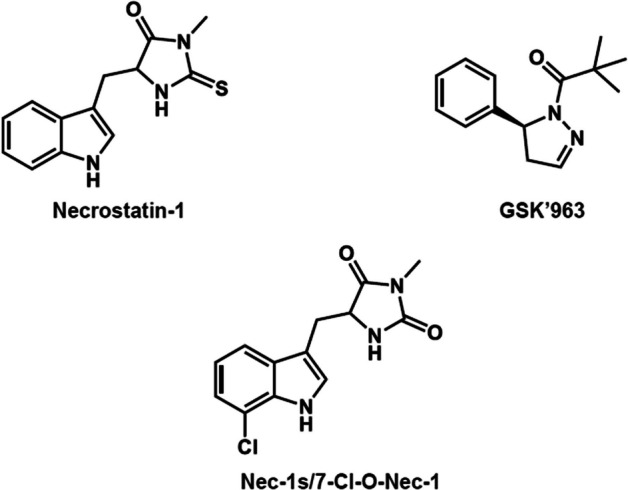
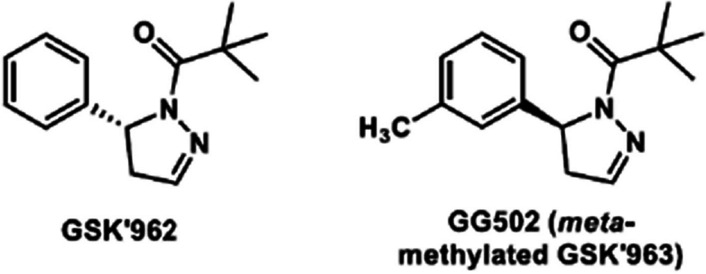
(S)-2,2-dimethyl-1-(5-phenyl-4,5-dihydro-1H-pyrazol-1-yl)propan-1-one (GSK’963) has a high affinity, selectivity for RIPK1, and favorable physiochemical properties based on its chemical structure. In this study, since 11C-labeling (half-life: 20.4 min) GSK’963 retaining its structure requiring the Grignard reaction of tert-butylmagnesium halides and [11C]carbon dioxide was anticipated to give a low yield, we decided instead to 11C-label a GSK’963 analog ((S)-2,2-dimethyl-1-(5-(m-tolyl)-4,5-dihydro-1H-pyrazol-1-yl)propan-1-one, GG502), which has a high RIPK1 inhibitory activity equivalent to that of the original compound GSK’963. Thus, we successfully 11C-labeled GG502 using a Pd-mediated cross-coupling reaction in favorable yields (3.6 ± 1.9%) and radiochemical purities (> 96%), and molar activity (47–115 GBq/μmol). On autoradiography, radioactivity accumulation was observed for [11C]GG502 and decreased by non-radioactive GG502 in the mouse spleen and human brain, indicating the possibility of specific binding of this ligand to RIPK1. On brain PET imaging in a rhesus monkey, [11C]GG502 showed a good brain permeability (peak standardized uptake value (SUV) ~3.0), although there was no clear evidence of specific binding of [11C]GG502. On brain PET imaging in acute inflammation model rats, [11C]GG502 also showed a good brain permeability, and no significant increased uptake was observed in the lipopolysaccharide-treated side of striatum. On metabolite analysis in rats at 30 min after administration of [11C]GG502, ~55% and ~10% of radioactivity was from unmetabolized [11C]GG502 in the brain and the plasma, respectively.
Conclusions
We synthesized and evaluated a 11C-labeled PET ligand based on the methylated analog of GSK’963 for imaging of RIPK1 in the brain. Although in autoradiography of the resulting [11C]GG502 indicated the possibility of specific binding, the actual PET imaging failed to detect any evidence of specific binding to RIPK1 despite its good brain permeability. Further development of radioligands with a higher binding affinity for RIPK1 in vivo and more stable metabolite profiles compared with the current compound may be required.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


