{"title":"NetMHCphosPan-Pan特异性预测磷酸化配体的MHC I类抗原呈递","authors":"Carina Thusgaard Refsgaard , Carolina Barra , Xu Peng , Nicola Ternette , Morten Nielsen","doi":"10.1016/j.immuno.2021.100005","DOIUrl":null,"url":null,"abstract":"<div><p>Post-translational modifications of proteins play a crucial part in carcinogenesis. Phosphorylated peptides have shown to be presented by MHC class I molecules and recognised by cytotoxic T cells, making them a promising target for immunotherapy. Identification of phosphorylated MHC class I ligands has so far predominantly been done using bioinformatic tools trained on unmodified peptides. Only one tool, PhosMHCpred, has been developed specifically for the prediction of phosphorylated MHC class I ligands so far and this tool has been trained only on a limited number of alleles and provides a limited peptide length coverage (only including 9-mers).</p><p>Here we propose a method, termed NetMHCphosPan, for the prediction of MHC presented phosphopeptides. The method is trained using the NNAlign_MA framework, which allows incorporating mixed data types and information leverage between data sets resulting in a greatly improved MHC and peptide length coverage and an overall increased predictive power compared to PhosMHCpred. Motif deconvolution suggested a strong preference for phosphosites to be located in position 4 of the binding motif, and enrichment of proline at P5 and arginine at P1. The improved performance, driven by the extended length and allelic coverage, of NetMHCphosPan over current state-of-the-art methods, was further validated on a large benchmark data set independent from the model development.</p><p>In conclusion, we have confirmed the high power of NNAlign_MA for motif deconvolution of complex immuno-peptidomics data and have developed a novel method for prediction of MHC presented phosphopeptides with improved predictive power and a broader peptide length and MHC coverage compared to current state-of-the-art methods. The developed method is available at <span>http://www.cbs.dtu.dk/services/NetMHCphosPan-1.0</span><svg><path></path></svg>.</p></div>","PeriodicalId":73343,"journal":{"name":"Immunoinformatics (Amsterdam, Netherlands)","volume":"1 ","pages":"Article 100005"},"PeriodicalIF":0.0000,"publicationDate":"2021-10-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1016/j.immuno.2021.100005","citationCount":"5","resultStr":"{\"title\":\"NetMHCphosPan - Pan-specific prediction of MHC class I antigen presentation of phosphorylated ligands\",\"authors\":\"Carina Thusgaard Refsgaard , Carolina Barra , Xu Peng , Nicola Ternette , Morten Nielsen\",\"doi\":\"10.1016/j.immuno.2021.100005\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>Post-translational modifications of proteins play a crucial part in carcinogenesis. Phosphorylated peptides have shown to be presented by MHC class I molecules and recognised by cytotoxic T cells, making them a promising target for immunotherapy. Identification of phosphorylated MHC class I ligands has so far predominantly been done using bioinformatic tools trained on unmodified peptides. Only one tool, PhosMHCpred, has been developed specifically for the prediction of phosphorylated MHC class I ligands so far and this tool has been trained only on a limited number of alleles and provides a limited peptide length coverage (only including 9-mers).</p><p>Here we propose a method, termed NetMHCphosPan, for the prediction of MHC presented phosphopeptides. The method is trained using the NNAlign_MA framework, which allows incorporating mixed data types and information leverage between data sets resulting in a greatly improved MHC and peptide length coverage and an overall increased predictive power compared to PhosMHCpred. Motif deconvolution suggested a strong preference for phosphosites to be located in position 4 of the binding motif, and enrichment of proline at P5 and arginine at P1. The improved performance, driven by the extended length and allelic coverage, of NetMHCphosPan over current state-of-the-art methods, was further validated on a large benchmark data set independent from the model development.</p><p>In conclusion, we have confirmed the high power of NNAlign_MA for motif deconvolution of complex immuno-peptidomics data and have developed a novel method for prediction of MHC presented phosphopeptides with improved predictive power and a broader peptide length and MHC coverage compared to current state-of-the-art methods. The developed method is available at <span>http://www.cbs.dtu.dk/services/NetMHCphosPan-1.0</span><svg><path></path></svg>.</p></div>\",\"PeriodicalId\":73343,\"journal\":{\"name\":\"Immunoinformatics (Amsterdam, Netherlands)\",\"volume\":\"1 \",\"pages\":\"Article 100005\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2021-10-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1016/j.immuno.2021.100005\",\"citationCount\":\"5\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Immunoinformatics (Amsterdam, Netherlands)\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S2667119021000057\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Immunoinformatics (Amsterdam, Netherlands)","FirstCategoryId":"1085","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2667119021000057","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
NetMHCphosPan - Pan-specific prediction of MHC class I antigen presentation of phosphorylated ligands
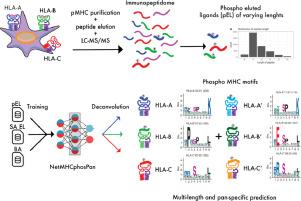
Post-translational modifications of proteins play a crucial part in carcinogenesis. Phosphorylated peptides have shown to be presented by MHC class I molecules and recognised by cytotoxic T cells, making them a promising target for immunotherapy. Identification of phosphorylated MHC class I ligands has so far predominantly been done using bioinformatic tools trained on unmodified peptides. Only one tool, PhosMHCpred, has been developed specifically for the prediction of phosphorylated MHC class I ligands so far and this tool has been trained only on a limited number of alleles and provides a limited peptide length coverage (only including 9-mers).
Here we propose a method, termed NetMHCphosPan, for the prediction of MHC presented phosphopeptides. The method is trained using the NNAlign_MA framework, which allows incorporating mixed data types and information leverage between data sets resulting in a greatly improved MHC and peptide length coverage and an overall increased predictive power compared to PhosMHCpred. Motif deconvolution suggested a strong preference for phosphosites to be located in position 4 of the binding motif, and enrichment of proline at P5 and arginine at P1. The improved performance, driven by the extended length and allelic coverage, of NetMHCphosPan over current state-of-the-art methods, was further validated on a large benchmark data set independent from the model development.
In conclusion, we have confirmed the high power of NNAlign_MA for motif deconvolution of complex immuno-peptidomics data and have developed a novel method for prediction of MHC presented phosphopeptides with improved predictive power and a broader peptide length and MHC coverage compared to current state-of-the-art methods. The developed method is available at http://www.cbs.dtu.dk/services/NetMHCphosPan-1.0.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


