{"title":"基于深度残差密集网络(DRDN)的改进蛋白质实值距离预测","authors":"S. Geethu, E. R. Vimina","doi":"10.1007/s10930-022-10067-4","DOIUrl":null,"url":null,"abstract":"<div><p>Three-dimensional protein structure prediction is one of the major challenges in bioinformatics. According to recent research findings, real-valued distance prediction plays a vital role in determining the unique three-dimensional protein structure. This paper proposes a novel methodology involving a deep residual dense network (DRDN) for predicting protein real-valued distance. The features extracted from the given query protein sequence and its corresponding homologous sequences are used for training the model. Multi-aligned homologous sequences for each query protein sequence are retrieved from five different databases using DeepMSA, HHblits, and HITS_PR_HHblits methods. The proposed method yielded outcomes of 3.89, 0.23, 0.45, and 0.63, respectively, corresponding to the evaluation metrics such as Absolute Error, Relative Error, High-accuracy Pairwise Distance Test (PDA), and Pairwise Distance Test (PDT). Further, the contact map is computed based on CASP criteria by converting the predicted real-valued distance, and it is evaluated using the precision metric. It is observed that precision of long-range top L/5 contact prediction on the CASP13 dataset by the proposed method, RaptorX, Zhang, trRosetta, JinboXu & JinLu, and Deepdist are 0.834, 0.657, 0.70, 0.785, 0.786, and 0.812, respectively. Also, Top-L/5 contact prediction on the CASP14 dataset evaluated using average precision resulted in 0.847, 0.707, 0.752, 0.783, 0.792, 0.817, and 0.825 respectively, corresponding to the proposed method, Zhang, RaptorX, trRosetta, Deepdist, JinboXu & JinLu, and Alphafold2.</p></div>","PeriodicalId":793,"journal":{"name":"The Protein Journal","volume":"41 4-5","pages":"468 - 476"},"PeriodicalIF":1.4000,"publicationDate":"2022-08-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://link.springer.com/content/pdf/10.1007/s10930-022-10067-4.pdf","citationCount":"1","resultStr":"{\"title\":\"Improved Protein Real-Valued Distance Prediction Using Deep Residual Dense Network (DRDN)\",\"authors\":\"S. Geethu, E. R. Vimina\",\"doi\":\"10.1007/s10930-022-10067-4\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>Three-dimensional protein structure prediction is one of the major challenges in bioinformatics. According to recent research findings, real-valued distance prediction plays a vital role in determining the unique three-dimensional protein structure. This paper proposes a novel methodology involving a deep residual dense network (DRDN) for predicting protein real-valued distance. The features extracted from the given query protein sequence and its corresponding homologous sequences are used for training the model. Multi-aligned homologous sequences for each query protein sequence are retrieved from five different databases using DeepMSA, HHblits, and HITS_PR_HHblits methods. The proposed method yielded outcomes of 3.89, 0.23, 0.45, and 0.63, respectively, corresponding to the evaluation metrics such as Absolute Error, Relative Error, High-accuracy Pairwise Distance Test (PDA), and Pairwise Distance Test (PDT). Further, the contact map is computed based on CASP criteria by converting the predicted real-valued distance, and it is evaluated using the precision metric. It is observed that precision of long-range top L/5 contact prediction on the CASP13 dataset by the proposed method, RaptorX, Zhang, trRosetta, JinboXu & JinLu, and Deepdist are 0.834, 0.657, 0.70, 0.785, 0.786, and 0.812, respectively. Also, Top-L/5 contact prediction on the CASP14 dataset evaluated using average precision resulted in 0.847, 0.707, 0.752, 0.783, 0.792, 0.817, and 0.825 respectively, corresponding to the proposed method, Zhang, RaptorX, trRosetta, Deepdist, JinboXu & JinLu, and Alphafold2.</p></div>\",\"PeriodicalId\":793,\"journal\":{\"name\":\"The Protein Journal\",\"volume\":\"41 4-5\",\"pages\":\"468 - 476\"},\"PeriodicalIF\":1.4000,\"publicationDate\":\"2022-08-25\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://link.springer.com/content/pdf/10.1007/s10930-022-10067-4.pdf\",\"citationCount\":\"1\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Protein Journal\",\"FirstCategoryId\":\"2\",\"ListUrlMain\":\"https://link.springer.com/article/10.1007/s10930-022-10067-4\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Protein Journal","FirstCategoryId":"2","ListUrlMain":"https://link.springer.com/article/10.1007/s10930-022-10067-4","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
Improved Protein Real-Valued Distance Prediction Using Deep Residual Dense Network (DRDN)
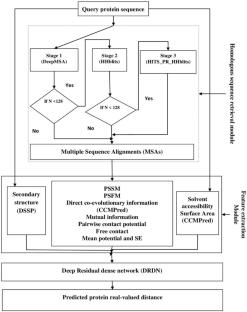
Three-dimensional protein structure prediction is one of the major challenges in bioinformatics. According to recent research findings, real-valued distance prediction plays a vital role in determining the unique three-dimensional protein structure. This paper proposes a novel methodology involving a deep residual dense network (DRDN) for predicting protein real-valued distance. The features extracted from the given query protein sequence and its corresponding homologous sequences are used for training the model. Multi-aligned homologous sequences for each query protein sequence are retrieved from five different databases using DeepMSA, HHblits, and HITS_PR_HHblits methods. The proposed method yielded outcomes of 3.89, 0.23, 0.45, and 0.63, respectively, corresponding to the evaluation metrics such as Absolute Error, Relative Error, High-accuracy Pairwise Distance Test (PDA), and Pairwise Distance Test (PDT). Further, the contact map is computed based on CASP criteria by converting the predicted real-valued distance, and it is evaluated using the precision metric. It is observed that precision of long-range top L/5 contact prediction on the CASP13 dataset by the proposed method, RaptorX, Zhang, trRosetta, JinboXu & JinLu, and Deepdist are 0.834, 0.657, 0.70, 0.785, 0.786, and 0.812, respectively. Also, Top-L/5 contact prediction on the CASP14 dataset evaluated using average precision resulted in 0.847, 0.707, 0.752, 0.783, 0.792, 0.817, and 0.825 respectively, corresponding to the proposed method, Zhang, RaptorX, trRosetta, Deepdist, JinboXu & JinLu, and Alphafold2.
期刊介绍:
The Protein Journal (formerly the Journal of Protein Chemistry) publishes original research work on all aspects of proteins and peptides. These include studies concerned with covalent or three-dimensional structure determination (X-ray, NMR, cryoEM, EPR/ESR, optical methods, etc.), computational aspects of protein structure and function, protein folding and misfolding, assembly, genetics, evolution, proteomics, molecular biology, protein engineering, protein nanotechnology, protein purification and analysis and peptide synthesis, as well as the elucidation and interpretation of the molecular bases of biological activities of proteins and peptides. We accept original research papers, reviews, mini-reviews, hypotheses, opinion papers, and letters to the editor.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


