{"title":"磷脂双层中n-烷烃的原子MD模拟:CHARMM36与Slipid","authors":"Anika Wurl, Tiago M. Ferreira","doi":"10.1002/mats.202200078","DOIUrl":null,"url":null,"abstract":"<p>Linear alkanes (<i>n</i>-alkanes) are chemically the most simple linear hydrophobic molecules in nature. Studying the incorporation of <i>n</i>-alkanes into lipid membranes is therefore a good starting point toward understanding the behavior of hydrophobic molecules in lipid membranes and to assess how accurately molecular dynamics models describe such systems. Here, the miscibility and structure of different <i>n</i>-alkanes—<i>n</i>-decane (C10), <i>n</i>-eicosane (C20), and <i>n</i>-triacontane (C30)—in dipalmitoylphosphatidylcholine membranes are investigated using two of the most used force fields for lipid membrane molecular dynamics simulations (CHARMM36 and Slipids). The <i>n</i>-alkanes are miscible in the membrane up to a critical volume fraction, ϕ<sub>c</sub>, that depends on the force field interaction parameters used. ϕ<sub>c</sub> is dependent on alkane chain length only for the model with more disordered chains (Slipids). Below ϕ<sub>c</sub>, a comparison with <sup>2</sup>H nuclear magnetic resonance (NMR) spectra indicates that a more realistic structure of the longer alkane molecules (C20 and C30) is obtained using the Slipids force field. On the other hand, for the shorter alkane (C10), Slipids simulations underestimate molecular order and CHARMM36 simulations enable a precise prediction of its experimental spectrum. The predicted <sup>2</sup>H NMR spectra are highly sensitive to 1–4 electrostatic interactions, and suggest that a reduction of the partial charges of the longer alkanes and acyl chains in CHARMM36 results in a better performance. The results presented indicate that lipid membranes with incorporated alkanes are highly valuable systems for the validation of force fields designed to perform lipid membrane simulations.</p>","PeriodicalId":18157,"journal":{"name":"Macromolecular Theory and Simulations","volume":"32 3","pages":""},"PeriodicalIF":1.8000,"publicationDate":"2023-03-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/mats.202200078","citationCount":"0","resultStr":"{\"title\":\"Atomistic MD Simulations of n-Alkanes in a Phospholipid Bilayer: CHARMM36 versus Slipids\",\"authors\":\"Anika Wurl, Tiago M. Ferreira\",\"doi\":\"10.1002/mats.202200078\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Linear alkanes (<i>n</i>-alkanes) are chemically the most simple linear hydrophobic molecules in nature. Studying the incorporation of <i>n</i>-alkanes into lipid membranes is therefore a good starting point toward understanding the behavior of hydrophobic molecules in lipid membranes and to assess how accurately molecular dynamics models describe such systems. Here, the miscibility and structure of different <i>n</i>-alkanes—<i>n</i>-decane (C10), <i>n</i>-eicosane (C20), and <i>n</i>-triacontane (C30)—in dipalmitoylphosphatidylcholine membranes are investigated using two of the most used force fields for lipid membrane molecular dynamics simulations (CHARMM36 and Slipids). The <i>n</i>-alkanes are miscible in the membrane up to a critical volume fraction, ϕ<sub>c</sub>, that depends on the force field interaction parameters used. ϕ<sub>c</sub> is dependent on alkane chain length only for the model with more disordered chains (Slipids). Below ϕ<sub>c</sub>, a comparison with <sup>2</sup>H nuclear magnetic resonance (NMR) spectra indicates that a more realistic structure of the longer alkane molecules (C20 and C30) is obtained using the Slipids force field. On the other hand, for the shorter alkane (C10), Slipids simulations underestimate molecular order and CHARMM36 simulations enable a precise prediction of its experimental spectrum. The predicted <sup>2</sup>H NMR spectra are highly sensitive to 1–4 electrostatic interactions, and suggest that a reduction of the partial charges of the longer alkanes and acyl chains in CHARMM36 results in a better performance. The results presented indicate that lipid membranes with incorporated alkanes are highly valuable systems for the validation of force fields designed to perform lipid membrane simulations.</p>\",\"PeriodicalId\":18157,\"journal\":{\"name\":\"Macromolecular Theory and Simulations\",\"volume\":\"32 3\",\"pages\":\"\"},\"PeriodicalIF\":1.8000,\"publicationDate\":\"2023-03-12\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/mats.202200078\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Macromolecular Theory and Simulations\",\"FirstCategoryId\":\"5\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/mats.202200078\",\"RegionNum\":4,\"RegionCategory\":\"工程技术\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"POLYMER SCIENCE\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Macromolecular Theory and Simulations","FirstCategoryId":"5","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/mats.202200078","RegionNum":4,"RegionCategory":"工程技术","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"POLYMER SCIENCE","Score":null,"Total":0}
Atomistic MD Simulations of n-Alkanes in a Phospholipid Bilayer: CHARMM36 versus Slipids
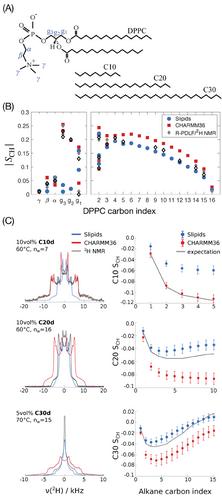
Linear alkanes (n-alkanes) are chemically the most simple linear hydrophobic molecules in nature. Studying the incorporation of n-alkanes into lipid membranes is therefore a good starting point toward understanding the behavior of hydrophobic molecules in lipid membranes and to assess how accurately molecular dynamics models describe such systems. Here, the miscibility and structure of different n-alkanes—n-decane (C10), n-eicosane (C20), and n-triacontane (C30)—in dipalmitoylphosphatidylcholine membranes are investigated using two of the most used force fields for lipid membrane molecular dynamics simulations (CHARMM36 and Slipids). The n-alkanes are miscible in the membrane up to a critical volume fraction, ϕc, that depends on the force field interaction parameters used. ϕc is dependent on alkane chain length only for the model with more disordered chains (Slipids). Below ϕc, a comparison with 2H nuclear magnetic resonance (NMR) spectra indicates that a more realistic structure of the longer alkane molecules (C20 and C30) is obtained using the Slipids force field. On the other hand, for the shorter alkane (C10), Slipids simulations underestimate molecular order and CHARMM36 simulations enable a precise prediction of its experimental spectrum. The predicted 2H NMR spectra are highly sensitive to 1–4 electrostatic interactions, and suggest that a reduction of the partial charges of the longer alkanes and acyl chains in CHARMM36 results in a better performance. The results presented indicate that lipid membranes with incorporated alkanes are highly valuable systems for the validation of force fields designed to perform lipid membrane simulations.
期刊介绍:
Macromolecular Theory and Simulations is the only high-quality polymer science journal dedicated exclusively to theory and simulations, covering all aspects from macromolecular theory to advanced computer simulation techniques.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


