Atanas Patronov, Ivan Dimitrov, Darren R Flower, Irini Doytchinova
{"title":"人类II类MHC等位基因HLA-DP2的肽结合预测:分子对接方法","authors":"Atanas Patronov, Ivan Dimitrov, Darren R Flower, Irini Doytchinova","doi":"10.1186/1472-6807-11-32","DOIUrl":null,"url":null,"abstract":"<p>MHC class II proteins bind oligopeptide fragments derived from proteolysis of pathogen antigens, presenting them at the cell surface for recognition by CD4+ T cells. Human MHC class II alleles are grouped into three loci: HLA-DP, HLA-DQ and HLA-DR. In contrast to HLA-DR and HLA-DQ, HLA-DP proteins have not been studied extensively, as they have been viewed as less important in immune responses than DRs and DQs. However, it is now known that HLA-DP alleles are associated with many autoimmune diseases. Quite recently, the X-ray structure of the HLA-DP2 molecule (DPA*0103, DPB1*0201) in complex with a self-peptide derived from the HLA-DR α-chain has been determined. In the present study, we applied a validated molecular docking protocol to a library of 247 modelled peptide-DP2 complexes, seeking to assess the contribution made by each of the 20 naturally occurred amino acids at each of the nine binding core peptide positions and the four flanking residues (two on both sides).</p><p>The free binding energies (FBEs) derived from the docking experiments were normalized on a position-dependent (npp) and on an overall basis (nap), and two docking score-based quantitative matrices (DS-QMs) were derived: QMnpp and QMnap. They reveal the amino acid preferences at each of the 13 positions considered in the study. Apart from the leading role of anchor positions p1 and p6, the binding to HLA-DP2 depends on the preferences at p2. No effect of the flanking residues was found on the peptide binding predictions to DP2, although all four of them show strong preferences for particular amino acids. The predictive ability of the DS-QMs was tested using a set of 457 known binders to HLA-DP2, originating from 24 proteins. The sensitivities of the predictions at five different thresholds (5%, 10%, 15%, 20% and 25%) were calculated and compared to the predictions made by the NetMHCII and IEDB servers. Analysis of the DS-QMs indicated an improvement in performance. Additionally, DS-QMs identified the binding cores of several known DP2 binders.</p><p>The molecular docking protocol, as applied to a combinatorial library of peptides, models the peptide-HLA-DP2 protein interaction effectively, generating reliable predictions in a quantitative assessment. The method is structure-based and does not require extensive experimental sequence-based data. Thus, it is universal and can be applied to model any peptide - protein interaction.</p>","PeriodicalId":498,"journal":{"name":"BMC Structural Biology","volume":"11 1","pages":""},"PeriodicalIF":2.2220,"publicationDate":"2011-07-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/1472-6807-11-32","citationCount":"58","resultStr":"{\"title\":\"Peptide binding prediction for the human class II MHC allele HLA-DP2: a molecular docking approach\",\"authors\":\"Atanas Patronov, Ivan Dimitrov, Darren R Flower, Irini Doytchinova\",\"doi\":\"10.1186/1472-6807-11-32\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>MHC class II proteins bind oligopeptide fragments derived from proteolysis of pathogen antigens, presenting them at the cell surface for recognition by CD4+ T cells. Human MHC class II alleles are grouped into three loci: HLA-DP, HLA-DQ and HLA-DR. In contrast to HLA-DR and HLA-DQ, HLA-DP proteins have not been studied extensively, as they have been viewed as less important in immune responses than DRs and DQs. However, it is now known that HLA-DP alleles are associated with many autoimmune diseases. Quite recently, the X-ray structure of the HLA-DP2 molecule (DPA*0103, DPB1*0201) in complex with a self-peptide derived from the HLA-DR α-chain has been determined. In the present study, we applied a validated molecular docking protocol to a library of 247 modelled peptide-DP2 complexes, seeking to assess the contribution made by each of the 20 naturally occurred amino acids at each of the nine binding core peptide positions and the four flanking residues (two on both sides).</p><p>The free binding energies (FBEs) derived from the docking experiments were normalized on a position-dependent (npp) and on an overall basis (nap), and two docking score-based quantitative matrices (DS-QMs) were derived: QMnpp and QMnap. They reveal the amino acid preferences at each of the 13 positions considered in the study. Apart from the leading role of anchor positions p1 and p6, the binding to HLA-DP2 depends on the preferences at p2. No effect of the flanking residues was found on the peptide binding predictions to DP2, although all four of them show strong preferences for particular amino acids. The predictive ability of the DS-QMs was tested using a set of 457 known binders to HLA-DP2, originating from 24 proteins. The sensitivities of the predictions at five different thresholds (5%, 10%, 15%, 20% and 25%) were calculated and compared to the predictions made by the NetMHCII and IEDB servers. Analysis of the DS-QMs indicated an improvement in performance. Additionally, DS-QMs identified the binding cores of several known DP2 binders.</p><p>The molecular docking protocol, as applied to a combinatorial library of peptides, models the peptide-HLA-DP2 protein interaction effectively, generating reliable predictions in a quantitative assessment. The method is structure-based and does not require extensive experimental sequence-based data. Thus, it is universal and can be applied to model any peptide - protein interaction.</p>\",\"PeriodicalId\":498,\"journal\":{\"name\":\"BMC Structural Biology\",\"volume\":\"11 1\",\"pages\":\"\"},\"PeriodicalIF\":2.2220,\"publicationDate\":\"2011-07-14\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/1472-6807-11-32\",\"citationCount\":\"58\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"BMC Structural Biology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://link.springer.com/article/10.1186/1472-6807-11-32\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"Biochemistry, Genetics and Molecular Biology\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Structural Biology","FirstCategoryId":"1085","ListUrlMain":"https://link.springer.com/article/10.1186/1472-6807-11-32","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
Peptide binding prediction for the human class II MHC allele HLA-DP2: a molecular docking approach
MHC class II proteins bind oligopeptide fragments derived from proteolysis of pathogen antigens, presenting them at the cell surface for recognition by CD4+ T cells. Human MHC class II alleles are grouped into three loci: HLA-DP, HLA-DQ and HLA-DR. In contrast to HLA-DR and HLA-DQ, HLA-DP proteins have not been studied extensively, as they have been viewed as less important in immune responses than DRs and DQs. However, it is now known that HLA-DP alleles are associated with many autoimmune diseases. Quite recently, the X-ray structure of the HLA-DP2 molecule (DPA*0103, DPB1*0201) in complex with a self-peptide derived from the HLA-DR α-chain has been determined. In the present study, we applied a validated molecular docking protocol to a library of 247 modelled peptide-DP2 complexes, seeking to assess the contribution made by each of the 20 naturally occurred amino acids at each of the nine binding core peptide positions and the four flanking residues (two on both sides).
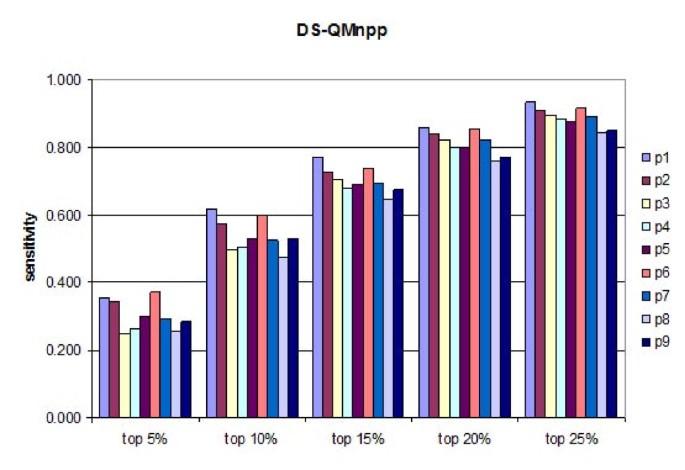
The free binding energies (FBEs) derived from the docking experiments were normalized on a position-dependent (npp) and on an overall basis (nap), and two docking score-based quantitative matrices (DS-QMs) were derived: QMnpp and QMnap. They reveal the amino acid preferences at each of the 13 positions considered in the study. Apart from the leading role of anchor positions p1 and p6, the binding to HLA-DP2 depends on the preferences at p2. No effect of the flanking residues was found on the peptide binding predictions to DP2, although all four of them show strong preferences for particular amino acids. The predictive ability of the DS-QMs was tested using a set of 457 known binders to HLA-DP2, originating from 24 proteins. The sensitivities of the predictions at five different thresholds (5%, 10%, 15%, 20% and 25%) were calculated and compared to the predictions made by the NetMHCII and IEDB servers. Analysis of the DS-QMs indicated an improvement in performance. Additionally, DS-QMs identified the binding cores of several known DP2 binders.
The molecular docking protocol, as applied to a combinatorial library of peptides, models the peptide-HLA-DP2 protein interaction effectively, generating reliable predictions in a quantitative assessment. The method is structure-based and does not require extensive experimental sequence-based data. Thus, it is universal and can be applied to model any peptide - protein interaction.
期刊介绍:
BMC Structural Biology is an open access, peer-reviewed journal that considers articles on investigations into the structure of biological macromolecules, including solving structures, structural and functional analyses, and computational modeling.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


