基于三分支多尺度卷积神经网络的药物靶点结合亲和力预测
IF 2.9
3区 生物学
Q3 BIOCHEMICAL RESEARCH METHODS
引用次数: 0
摘要
新药成本高、耗时长,而且往往伴随着安全问题。随着深度学习的发展,计算机辅助药物设计变得更加主流,卷积神经网络和图神经网络已被广泛用于药物-靶标亲和力(DTA)预测。本文提出了一种利用图卷积网络和多尺度卷积神经网络预测DTA的方法。我们将药物分子构建成图表示向量,并通过图注意力网络和图卷积网络学习特征表达。三分支卷积神经网络学习蛋白质序列的局部和全局特征,并将这两个特征表示合并到一个回归模块中以预测DTA。我们提出了一种预测DTA的新模型,与DeepDTA相比,Davis数据集的一致性指数提高了2.5%,均方误差的准确率提高了21%。此外,我们的方法优于其他主流DTA预测模型,即GANsDTA、WideDTA、GraphDTA和DeepAffinity。结果表明,在捕获蛋白质特征方面,使用多尺度卷积神经网络比使用单分支卷积神经网络更好,并且使用图来表达药物分子产生了更好的结果。本文章由计算机程序翻译,如有差异,请以英文原文为准。
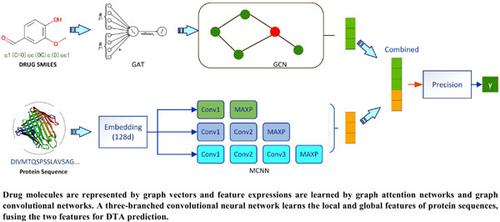
Drug–target binding affinity prediction based on three-branched multiscale convolutional neural networks
New drugs are costly, time-consuming, and often accompanied by safety concerns. With the development of deep learning, computer-aided drug design has become more mainstream, and convolutional neural networks and graph neural networks have been widely used for drug–target affinity (DTA) prediction.
The paper proposes a method of predicting DTA using graph convolutional networks and multiscale convolutional neural networks.
We construct drug molecules into graph representation vectors and learn feature expressions through graph attention networks and graph convolutional networks. A three-branch convolutional neural network learns the local and global features of protein sequences, and the two feature representations are merged into a regression module to predict the DTA.
We present a novel model to predict DTA, with a 2.5% improvement in the consistency index and a 21% accuracy improvement in terms of the mean squared error on the Davis dataset compared to DeepDTA. Morever, our method outperformed other mainstream DTA prediction models namely, GANsDTA, WideDTA, GraphDTA and DeepAffinity.
The results showed that the use of multiscale convolutional neural networks was better than a single-branched convolutional neural network at capturing protein signatures and the use of graphs to express drug molecules yielded better results.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Current Bioinformatics
生物-生化研究方法
CiteScore
6.60
自引率
2.50%
发文量
77
审稿时长
>12 weeks
期刊介绍:
Current Bioinformatics aims to publish all the latest and outstanding developments in bioinformatics. Each issue contains a series of timely, in-depth/mini-reviews, research papers and guest edited thematic issues written by leaders in the field, covering a wide range of the integration of biology with computer and information science.
The journal focuses on advances in computational molecular/structural biology, encompassing areas such as computing in biomedicine and genomics, computational proteomics and systems biology, and metabolic pathway engineering. Developments in these fields have direct implications on key issues related to health care, medicine, genetic disorders, development of agricultural products, renewable energy, environmental protection, etc.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


