Olivia Monteiro, Yan Wa Li, Daniel T. Baptista-Hon
{"title":"2022年猴痘病毒暴发的系统基因组特征——持续遗传监测的重要性","authors":"Olivia Monteiro, Yan Wa Li, Daniel T. Baptista-Hon","doi":"10.1002/mef2.16","DOIUrl":null,"url":null,"abstract":"<p>Monkeypox cases are steadily increasing worldwide. Phylogenetic characterization of the monkeypox virus (MPXV) responsible is important for epidemiological studies. Isidro et al. performed shotgun metagenomics sequencing analysis of MPXV isolated from cases in the current outbreak, published in <i>Nature Medicine</i>.<span><sup>1</sup></span> The results revealed for the first time that these samples cluster with a lower fatality clade of MPXV and that the current outbreak had a common origin. Furthermore, the study found evidence of accelerated evolution in the samples. This study highlights the importance of timely and sustained sequencing efforts to track the potential evolutionary trajectory of MPXV to mitigate its potential impact on global health.</p><p>The COVID-19 pandemic and other severe infectious disease outbreaks in recent years have in common among them the ability to cause infections across borders, likely facilitated by air travel and our changing relationship with rural areas.<span><sup>2</sup></span> The recovery of cross-country travel following the COVID-19 slowdown increases the possibility of bringing predominantly endemic infectious diseases to naïve populations. Reports of monkeypox around the world may be an example and potentially represents the next infectious disease challenge. The first case was recorded on May 6, 2022 in the United Kingdom.<span><sup>3</sup></span> As of June 27, 2022, over 4300 laboratory-confirmed monkeypox cases have been recorded across six continents.<span><sup>4</sup></span> We grouped daily new cases into weekly bins (Figure 1A), and it is clear that the number of cases is showing a persistent increase.</p><p>Monkeypox is caused by MPXV, a group of linear double-stranded DNA viruses part of the Orthopoxvirus genus.<span><sup>5</sup></span> Notable Orthopoxviruses include the Variola virus, the causative agent of smallpox and <i>Molluscum contagiosum</i>. All Orthopoxviruses are morphologically similar, with a brick-like structure (Figure 1B). The genome of Orthopoxviruses contains several hundred nonoverlapping open reading frames (ORFs). Many of these ORFs are highly conserved among members and are required for replication and morphogenesis. Others are divergent and result in heterogeneity in the host range, immune modulation properties, and pathogenesis. All Orthopoxviruses replicate in the cytoplasm of infected cells. Their life cycle is illustrated in Figure 1C.</p><p>Monkeypox has a variable incubation period (5–21 days), and is a self-limiting disease with symptoms lasting between 2 and 4 weeks.<span><sup>6</sup></span> The disease can be divided into two phases. The invasive prodromal phase is characterized by generalized systemic symptoms, such as fever, headache, lymphadenopathy, fatigue, and myalgia. The characteristic monkeypox rash is a feature of the cutaneous phase, which begins 1–3 days after fever onset. These are well-circumscribed, deep-seated, and painful maculopapular skin lesions, which typically occur on the face and then spread to the extremities. Complications of monkeypox are rare, and usually secondary to bronchopulmonary or corneal infections, leading to sepsis, encephalitis, and loss of vision.</p><p>MPXV is a zoonotic infection, endemic in more deprived communities in Central and Western Africa. MPXV can be separated into three clades<span><sup>6</sup></span>: Clade 1 (Central African or Congo Basin clade) and Clade 2/3 (collectively West African clade). Clade 1 has a case-fatality ratio (CFR) of 10%, while Clades 2 and 3 have a CFR of <1%. Before the current outbreak, cases of MPXV infections have known travel links to endemic areas or contact with infected animals. However, with the exception of the first confirmed case in the current outbreak (in the United Kingdom), most cases had not been to endemic areas, or contacted the index case. Furthermore, in the current outbreak cases of genital lesions occurring immediately after the prodromal phase have been reported in individuals with close contact with infected individuals, including men who have sex with men, highlighting a sexual route of infection.<span><sup>7</sup></span> This atypical epidemiology necessitates an analysis of the phylogenetic placement and evolutionary trends of the current MPXV outbreak. The phylogenomic analysis of MPXV reported by Isidro et al. is the first study to offer clues into the genetics of the MPXV strain responsible for the current outbreak.<span><sup>1</sup></span></p><p>This study is based on 15 clinical MPXV samples obtained from lesions and vesicle swabs. The samples were subjected to shotgun metagenomics sequencing and mapped to a reference genome (MPXV-UK_P2, 2018; GenBank accession #MT903344.1). A phylogenetic analysis was performed, using Zaire-96-I-16 (RefSeq accession #NC_003310.1) as a reference sequence for Clade 1 of the global phylogeny, which revealed that these MPXV samples belong to the less virulent Clade 3. Further analysis of other MPXV sequences from NCBI (until June 15, 2022) showed that the current outbreak cluster (lineage B.1.1) forms a divergent branch from lineage B.1, which caused a large outbreak in Nigeria in 2017/2018. Furthermore, the current outbreak cluster differed from the most recent cluster (outbreak in 2018/2019) by 50 single-nucleotide polymorphisms (SNPs). The authors speculated that the current MPXV cluster originated from the virus that caused the 2017/2018 outbreak in Nigeria and continued to evolve. The epidemiological finding of cases in multiple countries within 3 weeks of the index case in the UK suggests that there may be more than one single origin.</p><p>Orthopoxviruses generally accumulate 1–2 SNPs per site per year, which means that the current cluster has 6- to 12-fold more SNPs than predicted.<span><sup>8</sup></span> Isidro et al. suggested that this may be due to accelerated evolution and hypothesized that this may be mediated by human apolipoprotein B mRNA-editing catalytic polypeptide-like 3 (APOBEC3) enzymes, which edit viral genomes to inhibit their replication and infectivity. Microevolution analysis focused on GA > AA and TC > TT replacements, a signature of APOBEC3-mediated genome editing. They found 26 GA > AA and 15 TC > TT replacements in the current MPXV cluster, resulting in 24 non-synonymous, 18 synonymous, and 4 intergenic replacements. Interestingly, 2 (out of 15) of the clinical samples share a frameshift deletion in a gene that codes for an Ankyrin-rich host range protein, resulting in gene loss. The authors speculated that such gene loss during secondary transmission resulted from microevolution of the virus during human-to-human transmission. Additionally, the study also found non-synonymous intra-patient single nucleotide variants (iSNVs) in genes with known immune modulation functions in five of the samples, suggesting an evolutionary adaptation for efficient replication in humans.</p><p>The Isidro et al. study has provided the scientific and medical community the first glimpse into the phylogeny of the current MPXV outbreak. Promisingly, it has been identified that the phylogeny of the current outbreak belongs to the lower CFR Clade 3. However, it has also found evidence of accelerated evolution and genomic heterogeneity within single patients. These findings highlight the importance of continued genetic surveillance of the MPXV variants in the current outbreak. Many of the mutations found were non-synonymous, which may impact MPXV infectivity, transmissibility, host range, and also immune modulation. However, there are also a number of synonymous mutations, which may also impact MPXV function. This is particularly important in the context of recent findings that synonymous mutations are rarely neutral.<span><sup>9</sup></span> Therefore, there is a need to characterize these mutations in functional studies.</p><p>Upgrading the current outbreak to one of international concern may depend on the evolutionary trajectory of MPXV, perhaps toward higher virulence and/or CFR. Unmitigated spread increases the rate at which variants occur, and therefore protecting at-risk groups by vaccination will be a priority. A smallpox vaccine with efficacy against MPXV infection is being offered to at-risk groups in the United Kingdom.<span><sup>10</sup></span> Furthermore, Tecovirimat is a small-molecule antiviral that has been approved for treating monkeypox in Europe since January 2022. These are vital weapons in our arsenal against this emerging infectious disease threat, and may perhaps put the global medical and scientific community on a more solid footing to deal with MPXV. We also need to draw on our experience with Covid-19 and step up genetic surveillance on MPXV, as highlighted by the Isidro et al. study.</p><p>Olivia Monteiro, Yan Wa Li, and Daniel T. Baptista-Hon analyzed the MPXV incidence data and produced the figures, and drafted the manuscript. Daniel T. Baptista-Hon conceived the research and provided overall direction. All authors agreed on the content of the final manuscript.</p><p>Author Daniel T. Baptista-Hon is an editorial board member of <i>MedComm – Future Medicine</i>. Author Daniel T. Baptista-Hon was not involved in the journal's review of, or decisions related to, this manuscript. The remaining authors declare no conflict of interest.</p><p>Not applicable.</p>","PeriodicalId":74135,"journal":{"name":"MedComm - Future medicine","volume":"1 1","pages":""},"PeriodicalIF":0.0000,"publicationDate":"2022-08-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/mef2.16","citationCount":"1","resultStr":"{\"title\":\"Phylogenomic characterization of the 2022 outbreak of monkeypox virus—The importance of sustained genetic surveillance\",\"authors\":\"Olivia Monteiro, Yan Wa Li, Daniel T. Baptista-Hon\",\"doi\":\"10.1002/mef2.16\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Monkeypox cases are steadily increasing worldwide. Phylogenetic characterization of the monkeypox virus (MPXV) responsible is important for epidemiological studies. Isidro et al. performed shotgun metagenomics sequencing analysis of MPXV isolated from cases in the current outbreak, published in <i>Nature Medicine</i>.<span><sup>1</sup></span> The results revealed for the first time that these samples cluster with a lower fatality clade of MPXV and that the current outbreak had a common origin. Furthermore, the study found evidence of accelerated evolution in the samples. This study highlights the importance of timely and sustained sequencing efforts to track the potential evolutionary trajectory of MPXV to mitigate its potential impact on global health.</p><p>The COVID-19 pandemic and other severe infectious disease outbreaks in recent years have in common among them the ability to cause infections across borders, likely facilitated by air travel and our changing relationship with rural areas.<span><sup>2</sup></span> The recovery of cross-country travel following the COVID-19 slowdown increases the possibility of bringing predominantly endemic infectious diseases to naïve populations. Reports of monkeypox around the world may be an example and potentially represents the next infectious disease challenge. The first case was recorded on May 6, 2022 in the United Kingdom.<span><sup>3</sup></span> As of June 27, 2022, over 4300 laboratory-confirmed monkeypox cases have been recorded across six continents.<span><sup>4</sup></span> We grouped daily new cases into weekly bins (Figure 1A), and it is clear that the number of cases is showing a persistent increase.</p><p>Monkeypox is caused by MPXV, a group of linear double-stranded DNA viruses part of the Orthopoxvirus genus.<span><sup>5</sup></span> Notable Orthopoxviruses include the Variola virus, the causative agent of smallpox and <i>Molluscum contagiosum</i>. All Orthopoxviruses are morphologically similar, with a brick-like structure (Figure 1B). The genome of Orthopoxviruses contains several hundred nonoverlapping open reading frames (ORFs). Many of these ORFs are highly conserved among members and are required for replication and morphogenesis. Others are divergent and result in heterogeneity in the host range, immune modulation properties, and pathogenesis. All Orthopoxviruses replicate in the cytoplasm of infected cells. Their life cycle is illustrated in Figure 1C.</p><p>Monkeypox has a variable incubation period (5–21 days), and is a self-limiting disease with symptoms lasting between 2 and 4 weeks.<span><sup>6</sup></span> The disease can be divided into two phases. The invasive prodromal phase is characterized by generalized systemic symptoms, such as fever, headache, lymphadenopathy, fatigue, and myalgia. The characteristic monkeypox rash is a feature of the cutaneous phase, which begins 1–3 days after fever onset. These are well-circumscribed, deep-seated, and painful maculopapular skin lesions, which typically occur on the face and then spread to the extremities. Complications of monkeypox are rare, and usually secondary to bronchopulmonary or corneal infections, leading to sepsis, encephalitis, and loss of vision.</p><p>MPXV is a zoonotic infection, endemic in more deprived communities in Central and Western Africa. MPXV can be separated into three clades<span><sup>6</sup></span>: Clade 1 (Central African or Congo Basin clade) and Clade 2/3 (collectively West African clade). Clade 1 has a case-fatality ratio (CFR) of 10%, while Clades 2 and 3 have a CFR of <1%. Before the current outbreak, cases of MPXV infections have known travel links to endemic areas or contact with infected animals. However, with the exception of the first confirmed case in the current outbreak (in the United Kingdom), most cases had not been to endemic areas, or contacted the index case. Furthermore, in the current outbreak cases of genital lesions occurring immediately after the prodromal phase have been reported in individuals with close contact with infected individuals, including men who have sex with men, highlighting a sexual route of infection.<span><sup>7</sup></span> This atypical epidemiology necessitates an analysis of the phylogenetic placement and evolutionary trends of the current MPXV outbreak. The phylogenomic analysis of MPXV reported by Isidro et al. is the first study to offer clues into the genetics of the MPXV strain responsible for the current outbreak.<span><sup>1</sup></span></p><p>This study is based on 15 clinical MPXV samples obtained from lesions and vesicle swabs. The samples were subjected to shotgun metagenomics sequencing and mapped to a reference genome (MPXV-UK_P2, 2018; GenBank accession #MT903344.1). A phylogenetic analysis was performed, using Zaire-96-I-16 (RefSeq accession #NC_003310.1) as a reference sequence for Clade 1 of the global phylogeny, which revealed that these MPXV samples belong to the less virulent Clade 3. Further analysis of other MPXV sequences from NCBI (until June 15, 2022) showed that the current outbreak cluster (lineage B.1.1) forms a divergent branch from lineage B.1, which caused a large outbreak in Nigeria in 2017/2018. Furthermore, the current outbreak cluster differed from the most recent cluster (outbreak in 2018/2019) by 50 single-nucleotide polymorphisms (SNPs). The authors speculated that the current MPXV cluster originated from the virus that caused the 2017/2018 outbreak in Nigeria and continued to evolve. The epidemiological finding of cases in multiple countries within 3 weeks of the index case in the UK suggests that there may be more than one single origin.</p><p>Orthopoxviruses generally accumulate 1–2 SNPs per site per year, which means that the current cluster has 6- to 12-fold more SNPs than predicted.<span><sup>8</sup></span> Isidro et al. suggested that this may be due to accelerated evolution and hypothesized that this may be mediated by human apolipoprotein B mRNA-editing catalytic polypeptide-like 3 (APOBEC3) enzymes, which edit viral genomes to inhibit their replication and infectivity. Microevolution analysis focused on GA > AA and TC > TT replacements, a signature of APOBEC3-mediated genome editing. They found 26 GA > AA and 15 TC > TT replacements in the current MPXV cluster, resulting in 24 non-synonymous, 18 synonymous, and 4 intergenic replacements. Interestingly, 2 (out of 15) of the clinical samples share a frameshift deletion in a gene that codes for an Ankyrin-rich host range protein, resulting in gene loss. The authors speculated that such gene loss during secondary transmission resulted from microevolution of the virus during human-to-human transmission. Additionally, the study also found non-synonymous intra-patient single nucleotide variants (iSNVs) in genes with known immune modulation functions in five of the samples, suggesting an evolutionary adaptation for efficient replication in humans.</p><p>The Isidro et al. study has provided the scientific and medical community the first glimpse into the phylogeny of the current MPXV outbreak. Promisingly, it has been identified that the phylogeny of the current outbreak belongs to the lower CFR Clade 3. However, it has also found evidence of accelerated evolution and genomic heterogeneity within single patients. These findings highlight the importance of continued genetic surveillance of the MPXV variants in the current outbreak. Many of the mutations found were non-synonymous, which may impact MPXV infectivity, transmissibility, host range, and also immune modulation. However, there are also a number of synonymous mutations, which may also impact MPXV function. This is particularly important in the context of recent findings that synonymous mutations are rarely neutral.<span><sup>9</sup></span> Therefore, there is a need to characterize these mutations in functional studies.</p><p>Upgrading the current outbreak to one of international concern may depend on the evolutionary trajectory of MPXV, perhaps toward higher virulence and/or CFR. Unmitigated spread increases the rate at which variants occur, and therefore protecting at-risk groups by vaccination will be a priority. A smallpox vaccine with efficacy against MPXV infection is being offered to at-risk groups in the United Kingdom.<span><sup>10</sup></span> Furthermore, Tecovirimat is a small-molecule antiviral that has been approved for treating monkeypox in Europe since January 2022. These are vital weapons in our arsenal against this emerging infectious disease threat, and may perhaps put the global medical and scientific community on a more solid footing to deal with MPXV. We also need to draw on our experience with Covid-19 and step up genetic surveillance on MPXV, as highlighted by the Isidro et al. study.</p><p>Olivia Monteiro, Yan Wa Li, and Daniel T. Baptista-Hon analyzed the MPXV incidence data and produced the figures, and drafted the manuscript. Daniel T. Baptista-Hon conceived the research and provided overall direction. All authors agreed on the content of the final manuscript.</p><p>Author Daniel T. Baptista-Hon is an editorial board member of <i>MedComm – Future Medicine</i>. Author Daniel T. Baptista-Hon was not involved in the journal's review of, or decisions related to, this manuscript. The remaining authors declare no conflict of interest.</p><p>Not applicable.</p>\",\"PeriodicalId\":74135,\"journal\":{\"name\":\"MedComm - Future medicine\",\"volume\":\"1 1\",\"pages\":\"\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2022-08-16\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/mef2.16\",\"citationCount\":\"1\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"MedComm - Future medicine\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/mef2.16\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"MedComm - Future medicine","FirstCategoryId":"1085","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/mef2.16","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 1
摘要
全世界的猴痘病例正在稳步增加。猴痘病毒(MPXV)的系统发育特征对流行病学研究具有重要意义。Isidro等人对从当前疫情病例中分离的MPXV进行了散弹枪宏基因组测序分析,结果发表在《自然医学》(Nature medicine)杂志上。结果首次表明,这些样本与MPXV的低致死率分支聚集在一起,并且当前疫情具有共同的起源。此外,研究还在样本中发现了加速进化的证据。这项研究强调了及时和持续测序工作的重要性,以跟踪MPXV的潜在进化轨迹,以减轻其对全球健康的潜在影响。2019冠状病毒病大流行和近年来爆发的其他严重传染病有一个共同之处,即能够造成跨境感染,这可能是由于航空旅行和我们与农村地区关系的变化在COVID-19疫情放缓后,跨国旅行的复苏增加了将主要地方性传染病传播给naïve人群的可能性。世界各地关于猴痘的报告可能是一个例子,并可能代表下一个传染病挑战。第一例病例于2022年5月6日在英国被记录。截至2022年6月27日,六大洲已记录了4300多例实验室确诊猴痘病例我们将每天的新病例分组到每周的箱子中(图1A),很明显,病例数量正在持续增加。猴痘是由MPXV引起的,MPXV是一组线性双链DNA病毒,属于正痘病毒属著名的正痘病毒包括天花和传染性软疣的病原体天花病毒。所有正痘病毒在形态上相似,具有砖状结构(图1B)。正痘病毒的基因组包含数百个非重叠的开放阅读框(orf)。这些orf中的许多在成员之间高度保守,并且是复制和形态发生所必需的。其他的是不同的,导致宿主范围、免疫调节特性和发病机制的异质性。所有正痘病毒都在感染细胞的细胞质中复制。它们的生命周期如图1C所示。猴痘有不同的潜伏期(5-21天),是一种自限性疾病,症状持续2至4周该病可分为两个阶段。侵袭性前驱期的特点是全身性症状,如发热、头痛、淋巴结病、疲劳和肌痛。猴痘皮疹是皮肤期的特征,在发热后1-3天开始出现。这是一种界限分明、根深蒂固、疼痛的黄斑丘疹性皮肤病变,通常发生在面部,然后扩散到四肢。猴痘的并发症很少见,通常继发于支气管肺或角膜感染,导致败血症、脑炎和视力丧失。MPXV是一种人畜共患感染,在中非和西非较贫困的社区流行。MPXV可分为三个支系:支系1(中非或刚果盆地支系)和支系2/3(统称西非支系)。进化支1的病死率(CFR)为10%,而进化支2和3的病死率为1%。在当前疫情爆发之前,已知MPXV感染病例与流行地区有旅行联系或与受感染动物有接触。然而,除了本次疫情(在联合王国)中的第一例确诊病例外,大多数病例没有去过流行地区,也没有接触过指示病例。此外,在目前爆发的病例中,与感染者有密切接触的个人,包括男男性行为者,在前驱期后立即发生生殖器病变,突出了性感染途径这种非典型流行病学需要对当前MPXV暴发的系统发育定位和进化趋势进行分析。Isidro等人报道的MPXV系统发育分析是首次为导致当前疫情的MPXV毒株的遗传学提供线索的研究。本研究基于从病变和囊泡拭子中获得的15个临床MPXV样本。对样本进行散弹枪宏基因组测序,并将其定位到参考基因组(MPXV-UK_P2, 2018;GenBank序列号#MT903344.1)。采用扎伊尔-96- i -16 (RefSeq accession #NC_003310.1)作为全球系统发育分支1的参考序列进行系统发育分析,结果表明这些MPXV样本属于毒性较小的分支3。对NCBI其他MPXV序列的进一步分析(截至2022年6月15日)表明,目前的暴发聚集群(B.1.1谱系)与2017/2018年在尼日利亚引起大规模暴发的B.1谱系形成了不同的分支。 此外,当前爆发的聚集性与最近的聚集性(2018/2019年爆发)存在50个单核苷酸多态性(snp)的差异。作者推测,目前的MPXV集群源于导致2017/2018年尼日利亚疫情的病毒,并继续演变。在英国出现指示性病例后3周内,多个国家发现了病例,这表明可能存在不止一个传染源。正痘病毒通常每年在每个位点积累1-2个snp,这意味着目前的病毒群拥有比预期多6- 12倍的snpIsidro等人认为,这可能是由于加速进化,并假设这可能是由人类载脂蛋白B mrna编辑催化多肽样3 (APOBEC3)酶介导的,该酶编辑病毒基因组以抑制其复制和传染性。微进化分析的重点是GA > AA和TC > TT替换,这是apobec3介导的基因组编辑的标志。他们在当前的MPXV集群中发现了26个GA > AA和15个TC > TT替换,产生24个非同义替换,18个同义替换和4个基因间替换。有趣的是,15个临床样本中有2个在编码富含锚蛋白的宿主蛋白的基因中共享移码缺失,导致基因丢失。作者推测,这种基因在二次传播过程中的丢失是由于病毒在人际传播过程中的微进化造成的。此外,该研究还在五个样本中发现了具有已知免疫调节功能的基因中的非同义患者内单核苷酸变异(iSNVs),这表明进化适应了人类的有效复制。Isidro等人的研究为科学界和医学界提供了对当前MPXV暴发的系统发育的第一次了解。令人鼓舞的是,已经确定当前暴发的系统发育属于较低的CFR进化支3。然而,它也发现了在单个患者中加速进化和基因组异质性的证据。这些发现强调了在当前疫情中继续对MPXV变异体进行遗传监测的重要性。发现的许多突变是非同义的,这可能影响MPXV的传染性、传播性、宿主范围和免疫调节。然而,也有许多同义突变,它们也可能影响MPXV的功能。在最近发现同义突变很少是中性的背景下,这一点尤为重要因此,有必要在功能研究中描述这些突变。将当前疫情升级为国际关注的疫情可能取决于MPXV的进化轨迹,也许是朝着更高的毒力和/或CFR发展。未得到缓解的传播增加了变异发生的几率,因此通过接种疫苗保护高危人群将是一个优先事项。英国正在向高危人群提供一种对MPXV感染有效的天花疫苗。此外,Tecovirimat是一种小分子抗病毒药物,自2022年1月以来已在欧洲被批准用于治疗猴痘。这些都是我们应对这种新出现的传染病威胁的重要武器,也许会使全球医学和科学界在应对MPXV方面有更坚实的基础。正如Isidro等人的研究所强调的那样,我们还需要借鉴我们在Covid-19方面的经验,加强对MPXV的遗传监测。Olivia Monteiro, Yan Wa Li和Daniel T. Baptista-Hon分析了MPXV发病率数据并制作了这些数据,并起草了手稿。Daniel T. Baptista-Hon构思了这项研究并提供了总体指导。所有作者都同意最终稿的内容。作者Daniel T. Baptista-Hon是MedComm - Future Medicine的编辑委员会成员。作者Daniel T. Baptista-Hon没有参与该杂志对该手稿的审查或相关决定。其余作者声明没有利益冲突。不适用。
Phylogenomic characterization of the 2022 outbreak of monkeypox virus—The importance of sustained genetic surveillance
Monkeypox cases are steadily increasing worldwide. Phylogenetic characterization of the monkeypox virus (MPXV) responsible is important for epidemiological studies. Isidro et al. performed shotgun metagenomics sequencing analysis of MPXV isolated from cases in the current outbreak, published in Nature Medicine.1 The results revealed for the first time that these samples cluster with a lower fatality clade of MPXV and that the current outbreak had a common origin. Furthermore, the study found evidence of accelerated evolution in the samples. This study highlights the importance of timely and sustained sequencing efforts to track the potential evolutionary trajectory of MPXV to mitigate its potential impact on global health.
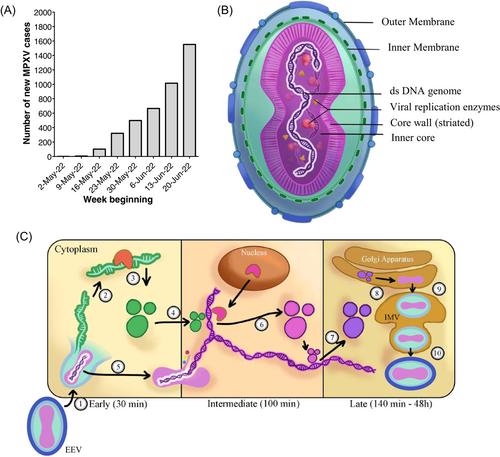
The COVID-19 pandemic and other severe infectious disease outbreaks in recent years have in common among them the ability to cause infections across borders, likely facilitated by air travel and our changing relationship with rural areas.2 The recovery of cross-country travel following the COVID-19 slowdown increases the possibility of bringing predominantly endemic infectious diseases to naïve populations. Reports of monkeypox around the world may be an example and potentially represents the next infectious disease challenge. The first case was recorded on May 6, 2022 in the United Kingdom.3 As of June 27, 2022, over 4300 laboratory-confirmed monkeypox cases have been recorded across six continents.4 We grouped daily new cases into weekly bins (Figure 1A), and it is clear that the number of cases is showing a persistent increase.
Monkeypox is caused by MPXV, a group of linear double-stranded DNA viruses part of the Orthopoxvirus genus.5 Notable Orthopoxviruses include the Variola virus, the causative agent of smallpox and Molluscum contagiosum. All Orthopoxviruses are morphologically similar, with a brick-like structure (Figure 1B). The genome of Orthopoxviruses contains several hundred nonoverlapping open reading frames (ORFs). Many of these ORFs are highly conserved among members and are required for replication and morphogenesis. Others are divergent and result in heterogeneity in the host range, immune modulation properties, and pathogenesis. All Orthopoxviruses replicate in the cytoplasm of infected cells. Their life cycle is illustrated in Figure 1C.
Monkeypox has a variable incubation period (5–21 days), and is a self-limiting disease with symptoms lasting between 2 and 4 weeks.6 The disease can be divided into two phases. The invasive prodromal phase is characterized by generalized systemic symptoms, such as fever, headache, lymphadenopathy, fatigue, and myalgia. The characteristic monkeypox rash is a feature of the cutaneous phase, which begins 1–3 days after fever onset. These are well-circumscribed, deep-seated, and painful maculopapular skin lesions, which typically occur on the face and then spread to the extremities. Complications of monkeypox are rare, and usually secondary to bronchopulmonary or corneal infections, leading to sepsis, encephalitis, and loss of vision.
MPXV is a zoonotic infection, endemic in more deprived communities in Central and Western Africa. MPXV can be separated into three clades6: Clade 1 (Central African or Congo Basin clade) and Clade 2/3 (collectively West African clade). Clade 1 has a case-fatality ratio (CFR) of 10%, while Clades 2 and 3 have a CFR of <1%. Before the current outbreak, cases of MPXV infections have known travel links to endemic areas or contact with infected animals. However, with the exception of the first confirmed case in the current outbreak (in the United Kingdom), most cases had not been to endemic areas, or contacted the index case. Furthermore, in the current outbreak cases of genital lesions occurring immediately after the prodromal phase have been reported in individuals with close contact with infected individuals, including men who have sex with men, highlighting a sexual route of infection.7 This atypical epidemiology necessitates an analysis of the phylogenetic placement and evolutionary trends of the current MPXV outbreak. The phylogenomic analysis of MPXV reported by Isidro et al. is the first study to offer clues into the genetics of the MPXV strain responsible for the current outbreak.1
This study is based on 15 clinical MPXV samples obtained from lesions and vesicle swabs. The samples were subjected to shotgun metagenomics sequencing and mapped to a reference genome (MPXV-UK_P2, 2018; GenBank accession #MT903344.1). A phylogenetic analysis was performed, using Zaire-96-I-16 (RefSeq accession #NC_003310.1) as a reference sequence for Clade 1 of the global phylogeny, which revealed that these MPXV samples belong to the less virulent Clade 3. Further analysis of other MPXV sequences from NCBI (until June 15, 2022) showed that the current outbreak cluster (lineage B.1.1) forms a divergent branch from lineage B.1, which caused a large outbreak in Nigeria in 2017/2018. Furthermore, the current outbreak cluster differed from the most recent cluster (outbreak in 2018/2019) by 50 single-nucleotide polymorphisms (SNPs). The authors speculated that the current MPXV cluster originated from the virus that caused the 2017/2018 outbreak in Nigeria and continued to evolve. The epidemiological finding of cases in multiple countries within 3 weeks of the index case in the UK suggests that there may be more than one single origin.
Orthopoxviruses generally accumulate 1–2 SNPs per site per year, which means that the current cluster has 6- to 12-fold more SNPs than predicted.8 Isidro et al. suggested that this may be due to accelerated evolution and hypothesized that this may be mediated by human apolipoprotein B mRNA-editing catalytic polypeptide-like 3 (APOBEC3) enzymes, which edit viral genomes to inhibit their replication and infectivity. Microevolution analysis focused on GA > AA and TC > TT replacements, a signature of APOBEC3-mediated genome editing. They found 26 GA > AA and 15 TC > TT replacements in the current MPXV cluster, resulting in 24 non-synonymous, 18 synonymous, and 4 intergenic replacements. Interestingly, 2 (out of 15) of the clinical samples share a frameshift deletion in a gene that codes for an Ankyrin-rich host range protein, resulting in gene loss. The authors speculated that such gene loss during secondary transmission resulted from microevolution of the virus during human-to-human transmission. Additionally, the study also found non-synonymous intra-patient single nucleotide variants (iSNVs) in genes with known immune modulation functions in five of the samples, suggesting an evolutionary adaptation for efficient replication in humans.
The Isidro et al. study has provided the scientific and medical community the first glimpse into the phylogeny of the current MPXV outbreak. Promisingly, it has been identified that the phylogeny of the current outbreak belongs to the lower CFR Clade 3. However, it has also found evidence of accelerated evolution and genomic heterogeneity within single patients. These findings highlight the importance of continued genetic surveillance of the MPXV variants in the current outbreak. Many of the mutations found were non-synonymous, which may impact MPXV infectivity, transmissibility, host range, and also immune modulation. However, there are also a number of synonymous mutations, which may also impact MPXV function. This is particularly important in the context of recent findings that synonymous mutations are rarely neutral.9 Therefore, there is a need to characterize these mutations in functional studies.
Upgrading the current outbreak to one of international concern may depend on the evolutionary trajectory of MPXV, perhaps toward higher virulence and/or CFR. Unmitigated spread increases the rate at which variants occur, and therefore protecting at-risk groups by vaccination will be a priority. A smallpox vaccine with efficacy against MPXV infection is being offered to at-risk groups in the United Kingdom.10 Furthermore, Tecovirimat is a small-molecule antiviral that has been approved for treating monkeypox in Europe since January 2022. These are vital weapons in our arsenal against this emerging infectious disease threat, and may perhaps put the global medical and scientific community on a more solid footing to deal with MPXV. We also need to draw on our experience with Covid-19 and step up genetic surveillance on MPXV, as highlighted by the Isidro et al. study.
Olivia Monteiro, Yan Wa Li, and Daniel T. Baptista-Hon analyzed the MPXV incidence data and produced the figures, and drafted the manuscript. Daniel T. Baptista-Hon conceived the research and provided overall direction. All authors agreed on the content of the final manuscript.
Author Daniel T. Baptista-Hon is an editorial board member of MedComm – Future Medicine. Author Daniel T. Baptista-Hon was not involved in the journal's review of, or decisions related to, this manuscript. The remaining authors declare no conflict of interest.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


