Arun Kumar Somavarapu, Satish Balakrishnan, Amit Kumar Singh Gautam, David S Palmer, Prasanna Venkatraman
{"title":"质谱法鉴定的磷蛋白质组结构分析揭示了允许和不允许的磷构象区域","authors":"Arun Kumar Somavarapu, Satish Balakrishnan, Amit Kumar Singh Gautam, David S Palmer, Prasanna Venkatraman","doi":"10.1186/1472-6807-14-9","DOIUrl":null,"url":null,"abstract":"<p>High-throughput mass spectrometric (HT-MS) study is the method of choice for monitoring global changes in proteome. Data derived from these studies are meant for further validation and experimentation to discover novel biological insights. Here we evaluate use of relative solvent accessible surface area (rSASA) and DEPTH as indices to assess experimentally determined phosphorylation events deposited in PhosphoSitePlus.</p><p>Based on accessibility, we map these identifications on allowed (accessible) or disallowed (inaccessible) regions of phosphoconformation. Surprisingly a striking number of HT-MS/MS derived events (1461/5947 sites or 24.6%) are present in the disallowed region of conformation. By considering protein dynamics, autophosphorylation events and/or the sequence specificity of kinases, 13.8% of these phosphosites can be moved to the allowed region of conformation. We also demonstrate that rSASA values can be used to increase the confidence of identification of phosphorylation sites within an ambiguous MS dataset.</p><p>While MS is a stand-alone technique for the identification of vast majority of phosphorylation events, identifications within disallowed region of conformation will benefit from techniques that independently probe for phosphorylation and protein dynamics. Our studies also imply that trapping alternate protein conformations may be a viable alternative to the design of inhibitors against mutation prone drug resistance kinases.</p>","PeriodicalId":498,"journal":{"name":"BMC Structural Biology","volume":"14 1","pages":""},"PeriodicalIF":2.2220,"publicationDate":"2014-03-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/1472-6807-14-9","citationCount":"9","resultStr":"{\"title\":\"Structural interrogation of phosphoproteome identified by mass spectrometry reveals allowed and disallowed regions of phosphoconformation\",\"authors\":\"Arun Kumar Somavarapu, Satish Balakrishnan, Amit Kumar Singh Gautam, David S Palmer, Prasanna Venkatraman\",\"doi\":\"10.1186/1472-6807-14-9\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>High-throughput mass spectrometric (HT-MS) study is the method of choice for monitoring global changes in proteome. Data derived from these studies are meant for further validation and experimentation to discover novel biological insights. Here we evaluate use of relative solvent accessible surface area (rSASA) and DEPTH as indices to assess experimentally determined phosphorylation events deposited in PhosphoSitePlus.</p><p>Based on accessibility, we map these identifications on allowed (accessible) or disallowed (inaccessible) regions of phosphoconformation. Surprisingly a striking number of HT-MS/MS derived events (1461/5947 sites or 24.6%) are present in the disallowed region of conformation. By considering protein dynamics, autophosphorylation events and/or the sequence specificity of kinases, 13.8% of these phosphosites can be moved to the allowed region of conformation. We also demonstrate that rSASA values can be used to increase the confidence of identification of phosphorylation sites within an ambiguous MS dataset.</p><p>While MS is a stand-alone technique for the identification of vast majority of phosphorylation events, identifications within disallowed region of conformation will benefit from techniques that independently probe for phosphorylation and protein dynamics. Our studies also imply that trapping alternate protein conformations may be a viable alternative to the design of inhibitors against mutation prone drug resistance kinases.</p>\",\"PeriodicalId\":498,\"journal\":{\"name\":\"BMC Structural Biology\",\"volume\":\"14 1\",\"pages\":\"\"},\"PeriodicalIF\":2.2220,\"publicationDate\":\"2014-03-11\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/1472-6807-14-9\",\"citationCount\":\"9\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"BMC Structural Biology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://link.springer.com/article/10.1186/1472-6807-14-9\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"Biochemistry, Genetics and Molecular Biology\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Structural Biology","FirstCategoryId":"1085","ListUrlMain":"https://link.springer.com/article/10.1186/1472-6807-14-9","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
Structural interrogation of phosphoproteome identified by mass spectrometry reveals allowed and disallowed regions of phosphoconformation
High-throughput mass spectrometric (HT-MS) study is the method of choice for monitoring global changes in proteome. Data derived from these studies are meant for further validation and experimentation to discover novel biological insights. Here we evaluate use of relative solvent accessible surface area (rSASA) and DEPTH as indices to assess experimentally determined phosphorylation events deposited in PhosphoSitePlus.
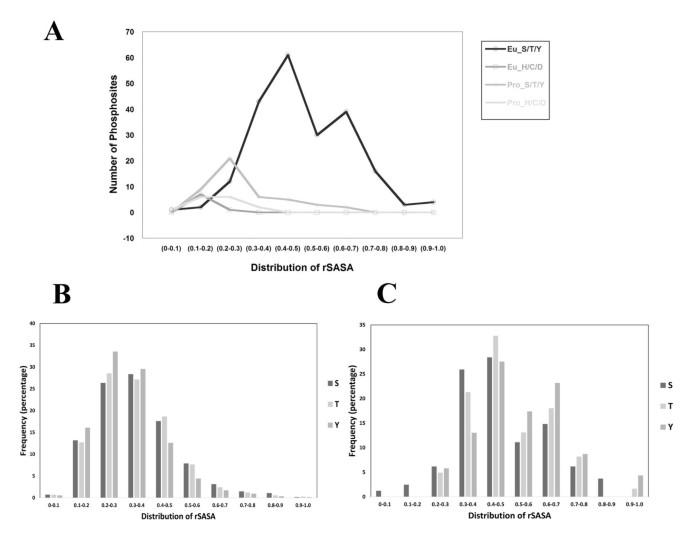
Based on accessibility, we map these identifications on allowed (accessible) or disallowed (inaccessible) regions of phosphoconformation. Surprisingly a striking number of HT-MS/MS derived events (1461/5947 sites or 24.6%) are present in the disallowed region of conformation. By considering protein dynamics, autophosphorylation events and/or the sequence specificity of kinases, 13.8% of these phosphosites can be moved to the allowed region of conformation. We also demonstrate that rSASA values can be used to increase the confidence of identification of phosphorylation sites within an ambiguous MS dataset.
While MS is a stand-alone technique for the identification of vast majority of phosphorylation events, identifications within disallowed region of conformation will benefit from techniques that independently probe for phosphorylation and protein dynamics. Our studies also imply that trapping alternate protein conformations may be a viable alternative to the design of inhibitors against mutation prone drug resistance kinases.
期刊介绍:
BMC Structural Biology is an open access, peer-reviewed journal that considers articles on investigations into the structure of biological macromolecules, including solving structures, structural and functional analyses, and computational modeling.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


