Morgan A. Bailey, Yun Tang, Hye-Jin Park and Michael C. Fitzgerald*,
{"title":"基于蛋白质折叠稳定性的生物学表型表征方法的比较分析","authors":"Morgan A. Bailey, Yun Tang, Hye-Jin Park and Michael C. Fitzgerald*, ","doi":"10.1021/jasms.2c00248","DOIUrl":null,"url":null,"abstract":"<p >Recently, a new suite of mass spectrometry-based proteomic methods has been developed that enables evaluation of protein folding stability on the proteomic scale. These methods utilize chemical and thermal denaturation approaches (SPROX and TPP, respectively) as well as proteolysis strategies (DARTS, LiP, and PP) to assess protein folding stability. The analytical capabilities of these technique have been well-established for protein target discovery applications. However, less is known about the relative advantages and disadvantages of using these different strategies to characterize biological phenotypes. Reported here is a comparative study of SPROX, TPP, LiP, and conventional protein expression level measurements using both a mouse model of aging and a mammalian cell culture model of breast cancer. Analyses on proteins in brain tissue cell lysates derived from 1- and 18-month-old mice (<i>n</i> = 4–5 at each time point) and on proteins in cell lysates derived from the MCF-7 and MCF-10A cell lines revealed a majority of the differentially stabilized protein hits in each phenotype analysis had unchanged expression levels. In both phenotype analyses, TPP generated the largest number and fraction of differentially stabilized protein hits. Only a quarter of all the protein hits identified in each phenotype analysis had a differential stability that was detected using multiple techniques. This work also reports the first peptide-level analysis of TPP data, which was required for the correct interpretation of the phenotype analyses performed here. Studies on selected protein stability hits also uncovered phenotype-related functional changes.</p>","PeriodicalId":672,"journal":{"name":"Journal of the American Society for Mass Spectrometry","volume":"34 3","pages":"383–393"},"PeriodicalIF":2.7000,"publicationDate":"2023-02-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.acs.org/doi/pdf/10.1021/jasms.2c00248","citationCount":"2","resultStr":"{\"title\":\"Comparative Analysis of Protein Folding Stability-Based Profiling Methods for Characterization of Biological Phenotypes\",\"authors\":\"Morgan A. Bailey, Yun Tang, Hye-Jin Park and Michael C. Fitzgerald*, \",\"doi\":\"10.1021/jasms.2c00248\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Recently, a new suite of mass spectrometry-based proteomic methods has been developed that enables evaluation of protein folding stability on the proteomic scale. These methods utilize chemical and thermal denaturation approaches (SPROX and TPP, respectively) as well as proteolysis strategies (DARTS, LiP, and PP) to assess protein folding stability. The analytical capabilities of these technique have been well-established for protein target discovery applications. However, less is known about the relative advantages and disadvantages of using these different strategies to characterize biological phenotypes. Reported here is a comparative study of SPROX, TPP, LiP, and conventional protein expression level measurements using both a mouse model of aging and a mammalian cell culture model of breast cancer. Analyses on proteins in brain tissue cell lysates derived from 1- and 18-month-old mice (<i>n</i> = 4–5 at each time point) and on proteins in cell lysates derived from the MCF-7 and MCF-10A cell lines revealed a majority of the differentially stabilized protein hits in each phenotype analysis had unchanged expression levels. In both phenotype analyses, TPP generated the largest number and fraction of differentially stabilized protein hits. Only a quarter of all the protein hits identified in each phenotype analysis had a differential stability that was detected using multiple techniques. This work also reports the first peptide-level analysis of TPP data, which was required for the correct interpretation of the phenotype analyses performed here. Studies on selected protein stability hits also uncovered phenotype-related functional changes.</p>\",\"PeriodicalId\":672,\"journal\":{\"name\":\"Journal of the American Society for Mass Spectrometry\",\"volume\":\"34 3\",\"pages\":\"383–393\"},\"PeriodicalIF\":2.7000,\"publicationDate\":\"2023-02-20\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://pubs.acs.org/doi/pdf/10.1021/jasms.2c00248\",\"citationCount\":\"2\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of the American Society for Mass Spectrometry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/jasms.2c00248\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of the American Society for Mass Spectrometry","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/jasms.2c00248","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
Comparative Analysis of Protein Folding Stability-Based Profiling Methods for Characterization of Biological Phenotypes
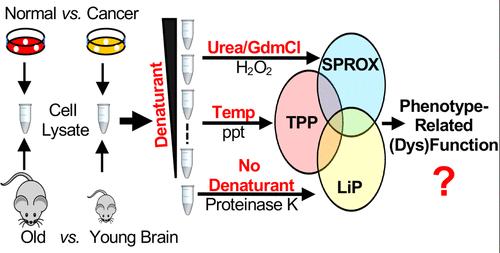
Recently, a new suite of mass spectrometry-based proteomic methods has been developed that enables evaluation of protein folding stability on the proteomic scale. These methods utilize chemical and thermal denaturation approaches (SPROX and TPP, respectively) as well as proteolysis strategies (DARTS, LiP, and PP) to assess protein folding stability. The analytical capabilities of these technique have been well-established for protein target discovery applications. However, less is known about the relative advantages and disadvantages of using these different strategies to characterize biological phenotypes. Reported here is a comparative study of SPROX, TPP, LiP, and conventional protein expression level measurements using both a mouse model of aging and a mammalian cell culture model of breast cancer. Analyses on proteins in brain tissue cell lysates derived from 1- and 18-month-old mice (n = 4–5 at each time point) and on proteins in cell lysates derived from the MCF-7 and MCF-10A cell lines revealed a majority of the differentially stabilized protein hits in each phenotype analysis had unchanged expression levels. In both phenotype analyses, TPP generated the largest number and fraction of differentially stabilized protein hits. Only a quarter of all the protein hits identified in each phenotype analysis had a differential stability that was detected using multiple techniques. This work also reports the first peptide-level analysis of TPP data, which was required for the correct interpretation of the phenotype analyses performed here. Studies on selected protein stability hits also uncovered phenotype-related functional changes.
期刊介绍:
The Journal of the American Society for Mass Spectrometry presents research papers covering all aspects of mass spectrometry, incorporating coverage of fields of scientific inquiry in which mass spectrometry can play a role.
Comprehensive in scope, the journal publishes papers on both fundamentals and applications of mass spectrometry. Fundamental subjects include instrumentation principles, design, and demonstration, structures and chemical properties of gas-phase ions, studies of thermodynamic properties, ion spectroscopy, chemical kinetics, mechanisms of ionization, theories of ion fragmentation, cluster ions, and potential energy surfaces. In addition to full papers, the journal offers Communications, Application Notes, and Accounts and Perspectives

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


