{"title":"TLK1B介导的Rad9磷酸化调节其核/细胞质定位和细胞周期检查点","authors":"Sanket Awate, Arrigo De Benedetti","doi":"10.1186/s12867-016-0056-x","DOIUrl":null,"url":null,"abstract":"<p>The Tousled like kinase 1B (TLK1B) is critical for DNA repair and survival of cells. Upon DNA damage, Chk1 phosphorylates TLK1B at S457 leading to its transient inhibition. Once TLK1B regains its kinase activity it phosphorylates Rad9 at S328. In this work we investigated the significance of this mechanism by overexpressing mutant TLK1B in which the inhibitory phosphorylation site was eliminated.</p><p>These cells expressing TLK1B resistant to DNA damage showed constitutive phosphorylation of Rad9 S328 that occurred even in the presence of hydroxyurea (HU), and this resulted in a delayed checkpoint recovery. One possible explanation was that premature phosphorylation of Rad9 caused its dissociation from 9-1-1 at stalled replication forks, resulting in their collapse and prolonged activation of the S-phase checkpoint. We found that phosphorylation of Rad9 at S328 results in its dissociation from chromatin and redistribution to the cytoplasm. This results in double stranded breaks formation with concomitant activation of ATM and phosphorylation of H2AX. Furthermore, a Rad9 (S328D) phosphomimic mutant was exclusively localized to the cytoplasm and not the chromatin. Another Rad9 phosphomimic mutant (T355D), which is also a site phosphorylated by TLK1, localized normally. In cells expressing the mutant TLK1B treated with HU, Rad9 association with Hus1 and WRN was greatly reduced, suggesting again that its phosphorylation causes its premature release from stalled forks.</p><p>We propose that normally, the inactivation of TLK1B following replication arrest and genotoxic stress functions to allow the retention of 9-1-1 at the sites of damage or stalled forks. Following reactivation of TLK1B, whose synthesis is concomitantly induced by genotoxins, Rad9 is hyperphosphorylated at S328, resulting in its dissociation and inactivation of the checkpoint that occurs once repair is complete.</p>","PeriodicalId":497,"journal":{"name":"BMC Molecular Biology","volume":"17 1","pages":""},"PeriodicalIF":2.9460,"publicationDate":"2016-02-09","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s12867-016-0056-x","citationCount":"12","resultStr":"{\"title\":\"TLK1B mediated phosphorylation of Rad9 regulates its nuclear/cytoplasmic localization and cell cycle checkpoint\",\"authors\":\"Sanket Awate, Arrigo De Benedetti\",\"doi\":\"10.1186/s12867-016-0056-x\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>The Tousled like kinase 1B (TLK1B) is critical for DNA repair and survival of cells. Upon DNA damage, Chk1 phosphorylates TLK1B at S457 leading to its transient inhibition. Once TLK1B regains its kinase activity it phosphorylates Rad9 at S328. In this work we investigated the significance of this mechanism by overexpressing mutant TLK1B in which the inhibitory phosphorylation site was eliminated.</p><p>These cells expressing TLK1B resistant to DNA damage showed constitutive phosphorylation of Rad9 S328 that occurred even in the presence of hydroxyurea (HU), and this resulted in a delayed checkpoint recovery. One possible explanation was that premature phosphorylation of Rad9 caused its dissociation from 9-1-1 at stalled replication forks, resulting in their collapse and prolonged activation of the S-phase checkpoint. We found that phosphorylation of Rad9 at S328 results in its dissociation from chromatin and redistribution to the cytoplasm. This results in double stranded breaks formation with concomitant activation of ATM and phosphorylation of H2AX. Furthermore, a Rad9 (S328D) phosphomimic mutant was exclusively localized to the cytoplasm and not the chromatin. Another Rad9 phosphomimic mutant (T355D), which is also a site phosphorylated by TLK1, localized normally. In cells expressing the mutant TLK1B treated with HU, Rad9 association with Hus1 and WRN was greatly reduced, suggesting again that its phosphorylation causes its premature release from stalled forks.</p><p>We propose that normally, the inactivation of TLK1B following replication arrest and genotoxic stress functions to allow the retention of 9-1-1 at the sites of damage or stalled forks. Following reactivation of TLK1B, whose synthesis is concomitantly induced by genotoxins, Rad9 is hyperphosphorylated at S328, resulting in its dissociation and inactivation of the checkpoint that occurs once repair is complete.</p>\",\"PeriodicalId\":497,\"journal\":{\"name\":\"BMC Molecular Biology\",\"volume\":\"17 1\",\"pages\":\"\"},\"PeriodicalIF\":2.9460,\"publicationDate\":\"2016-02-09\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/s12867-016-0056-x\",\"citationCount\":\"12\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"BMC Molecular Biology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://link.springer.com/article/10.1186/s12867-016-0056-x\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"Biochemistry, Genetics and Molecular Biology\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Molecular Biology","FirstCategoryId":"1085","ListUrlMain":"https://link.springer.com/article/10.1186/s12867-016-0056-x","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
引用次数: 12
摘要
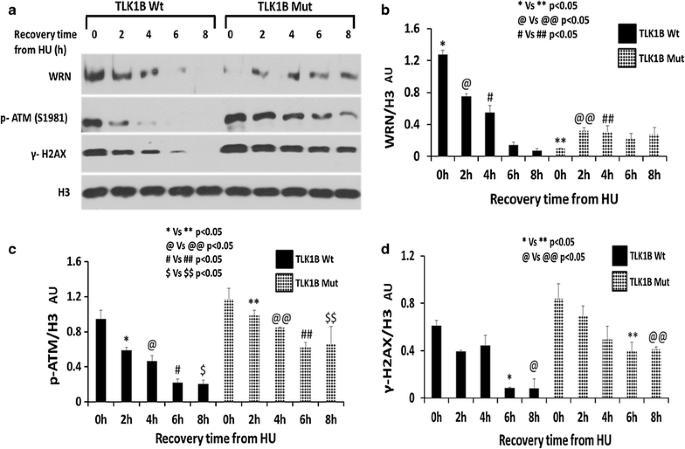
TLK1B激酶(Tousled like kinase 1B, TLK1B)对DNA修复和细胞存活至关重要。DNA损伤后,Chk1使TLK1B在S457位点磷酸化,导致其短暂抑制。一旦TLK1B恢复其激酶活性,它就会在S328位点磷酸化Rad9。在这项工作中,我们通过过表达抑制磷酸化位点被消除的突变体TLK1B来研究这种机制的意义。这些表达TLK1B抵抗DNA损伤的细胞显示出Rad9 S328的组成性磷酸化,即使在羟基脲(HU)存在的情况下也会发生,这导致检查点恢复延迟。一种可能的解释是,Rad9的过早磷酸化导致其在停滞的复制叉上与9-1-1分离,导致它们的崩溃和s期检查点的延长激活。我们发现Rad9在S328位点的磷酸化导致其与染色质分离并重新分布到细胞质中。这导致双链断裂形成,同时激活ATM和磷酸化H2AX。此外,Rad9 (S328D)磷酰亚胺突变体仅定位于细胞质而不是染色质。另一个Rad9缺磷突变体(T355D),也是一个被TLK1磷酸化的位点,正常定位。在表达TLK1B突变体的细胞中,经HU处理后,Rad9与Hus1和WRN的关联大大降低,再次表明其磷酸化导致其从停滞分叉中过早释放。我们认为,通常情况下,TLK1B在复制停滞和基因毒性应激后的失活功能允许9-1-1保留在损伤或停止分叉的部位。TLK1B的合成是由基因毒素诱导的,在TLK1B的再激活后,Rad9在S328位点被过度磷酸化,导致其解离并在修复完成后发生检查点失活。
TLK1B mediated phosphorylation of Rad9 regulates its nuclear/cytoplasmic localization and cell cycle checkpoint
The Tousled like kinase 1B (TLK1B) is critical for DNA repair and survival of cells. Upon DNA damage, Chk1 phosphorylates TLK1B at S457 leading to its transient inhibition. Once TLK1B regains its kinase activity it phosphorylates Rad9 at S328. In this work we investigated the significance of this mechanism by overexpressing mutant TLK1B in which the inhibitory phosphorylation site was eliminated.
These cells expressing TLK1B resistant to DNA damage showed constitutive phosphorylation of Rad9 S328 that occurred even in the presence of hydroxyurea (HU), and this resulted in a delayed checkpoint recovery. One possible explanation was that premature phosphorylation of Rad9 caused its dissociation from 9-1-1 at stalled replication forks, resulting in their collapse and prolonged activation of the S-phase checkpoint. We found that phosphorylation of Rad9 at S328 results in its dissociation from chromatin and redistribution to the cytoplasm. This results in double stranded breaks formation with concomitant activation of ATM and phosphorylation of H2AX. Furthermore, a Rad9 (S328D) phosphomimic mutant was exclusively localized to the cytoplasm and not the chromatin. Another Rad9 phosphomimic mutant (T355D), which is also a site phosphorylated by TLK1, localized normally. In cells expressing the mutant TLK1B treated with HU, Rad9 association with Hus1 and WRN was greatly reduced, suggesting again that its phosphorylation causes its premature release from stalled forks.
We propose that normally, the inactivation of TLK1B following replication arrest and genotoxic stress functions to allow the retention of 9-1-1 at the sites of damage or stalled forks. Following reactivation of TLK1B, whose synthesis is concomitantly induced by genotoxins, Rad9 is hyperphosphorylated at S328, resulting in its dissociation and inactivation of the checkpoint that occurs once repair is complete.
期刊介绍:
BMC Molecular Biology is an open access journal publishing original peer-reviewed research articles in all aspects of DNA and RNA in a cellular context, encompassing investigations of chromatin, replication, recombination, mutation, repair, transcription, translation and RNA processing and function.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


