Bobby Solanki, Pooja Sharma, Prabhat Ranjan, Pancham Kumar, Tanmoy Chakraborty
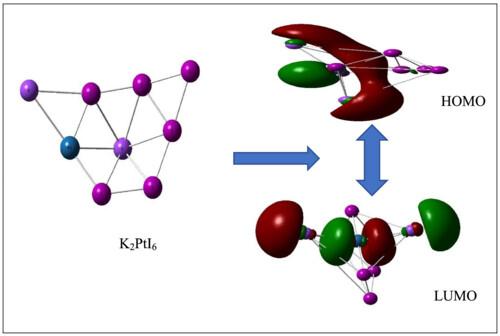
{"title":"双钙钛矿a2bi6 (A = Cs, K, Rb)的计算研究B = Pt, Sn)调用密度泛函理论","authors":"Bobby Solanki, Pooja Sharma, Prabhat Ranjan, Pancham Kumar, Tanmoy Chakraborty","doi":"10.1002/poc.4519","DOIUrl":null,"url":null,"abstract":"<p>Lead-free double perovskite materials A<sub>2</sub>BI<sub>6</sub> (A = Cs, K, Rb; B = Pt and Sn) have been studied and analyzed invoking density functional theory (DFT). Computed values of the HOMO–LUMO gap for lead-free double perovskites material A<sub>2</sub>BI<sub>6</sub> are found in the range of 1.062–2.811 eV. The energy gaps of K<sub>2</sub>PtI<sub>6</sub>, K<sub>2</sub>SnI<sub>6</sub>, and Rb<sub>2</sub>SnI<sub>6</sub> are in the optimal energy gap range (0.9 to 1.6 eV) required for a lead-free double perovskite system. Conceptual DFT-based descriptors, viz., molecular hardness, softness, electronegativity, electrophilicity index, dipole moment, and polarizability, are computed. The result reveals that K<sub>2</sub>PtI<sub>6</sub> shows high efficacy towards electron injection and may show the maximum electron driving force. The optical properties—refractive index and dielectric constant—of these perovskites are also computed. The maximum value of refractive index and dielectric constant is found for K<sub>2</sub>PtI<sub>6</sub>. Our computed results are in good agreement with the available experimental and other theoretical data. Perovskite materials K<sub>2</sub>PtI<sub>6</sub>, K<sub>2</sub>SnI<sub>6</sub>, and Rb<sub>2</sub>SnI<sub>6</sub> display a suitable energy gap as well as a high refractive index and dielectric constant, which makes them suitable for photovoltaic applications.</p>","PeriodicalId":16829,"journal":{"name":"Journal of Physical Organic Chemistry","volume":"36 12","pages":""},"PeriodicalIF":1.9000,"publicationDate":"2023-05-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"2","resultStr":"{\"title\":\"A computational study of double perovskites A2BI6 (A = Cs, K, Rb; B = Pt, Sn) invoking density functional theory\",\"authors\":\"Bobby Solanki, Pooja Sharma, Prabhat Ranjan, Pancham Kumar, Tanmoy Chakraborty\",\"doi\":\"10.1002/poc.4519\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Lead-free double perovskite materials A<sub>2</sub>BI<sub>6</sub> (A = Cs, K, Rb; B = Pt and Sn) have been studied and analyzed invoking density functional theory (DFT). Computed values of the HOMO–LUMO gap for lead-free double perovskites material A<sub>2</sub>BI<sub>6</sub> are found in the range of 1.062–2.811 eV. The energy gaps of K<sub>2</sub>PtI<sub>6</sub>, K<sub>2</sub>SnI<sub>6</sub>, and Rb<sub>2</sub>SnI<sub>6</sub> are in the optimal energy gap range (0.9 to 1.6 eV) required for a lead-free double perovskite system. Conceptual DFT-based descriptors, viz., molecular hardness, softness, electronegativity, electrophilicity index, dipole moment, and polarizability, are computed. The result reveals that K<sub>2</sub>PtI<sub>6</sub> shows high efficacy towards electron injection and may show the maximum electron driving force. The optical properties—refractive index and dielectric constant—of these perovskites are also computed. The maximum value of refractive index and dielectric constant is found for K<sub>2</sub>PtI<sub>6</sub>. Our computed results are in good agreement with the available experimental and other theoretical data. Perovskite materials K<sub>2</sub>PtI<sub>6</sub>, K<sub>2</sub>SnI<sub>6</sub>, and Rb<sub>2</sub>SnI<sub>6</sub> display a suitable energy gap as well as a high refractive index and dielectric constant, which makes them suitable for photovoltaic applications.</p>\",\"PeriodicalId\":16829,\"journal\":{\"name\":\"Journal of Physical Organic Chemistry\",\"volume\":\"36 12\",\"pages\":\"\"},\"PeriodicalIF\":1.9000,\"publicationDate\":\"2023-05-07\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"2\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Physical Organic Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/poc.4519\",\"RegionNum\":4,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, ORGANIC\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Physical Organic Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/poc.4519","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, ORGANIC","Score":null,"Total":0}
A computational study of double perovskites A2BI6 (A = Cs, K, Rb; B = Pt, Sn) invoking density functional theory
Lead-free double perovskite materials A2BI6 (A = Cs, K, Rb; B = Pt and Sn) have been studied and analyzed invoking density functional theory (DFT). Computed values of the HOMO–LUMO gap for lead-free double perovskites material A2BI6 are found in the range of 1.062–2.811 eV. The energy gaps of K2PtI6, K2SnI6, and Rb2SnI6 are in the optimal energy gap range (0.9 to 1.6 eV) required for a lead-free double perovskite system. Conceptual DFT-based descriptors, viz., molecular hardness, softness, electronegativity, electrophilicity index, dipole moment, and polarizability, are computed. The result reveals that K2PtI6 shows high efficacy towards electron injection and may show the maximum electron driving force. The optical properties—refractive index and dielectric constant—of these perovskites are also computed. The maximum value of refractive index and dielectric constant is found for K2PtI6. Our computed results are in good agreement with the available experimental and other theoretical data. Perovskite materials K2PtI6, K2SnI6, and Rb2SnI6 display a suitable energy gap as well as a high refractive index and dielectric constant, which makes them suitable for photovoltaic applications.
期刊介绍:
The Journal of Physical Organic Chemistry is the foremost international journal devoted to the relationship between molecular structure and chemical reactivity in organic systems. It publishes Research Articles, Reviews and Mini Reviews based on research striving to understand the principles governing chemical structures in relation to activity and transformation with physical and mathematical rigor, using results derived from experimental and computational methods. Physical Organic Chemistry is a central and fundamental field with multiple applications in fields such as molecular recognition, supramolecular chemistry, catalysis, photochemistry, biological and material sciences, nanotechnology and surface science.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


