{"title":"通过使用基因组序列的无比对机器学习方法探索人类适应冠状病毒的致命性","authors":"Rui Yin, Zihan Luo, Chee Keong Kwoh","doi":"10.2174/1389202923666211221110857","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>A newly emerging novel coronavirus appeared and rapidly spread worldwide and World Health Organization declared a pandemic on March 11, 2020. The roles and characteristics of coronavirus have captured much attention due to its power of causing a wide variety of infectious diseases, from mild to severe, on humans. The detection of the lethality of human coronavirus is key to estimate the viral toxicity and provide perspectives for treatment.</p><p><strong>Methods: </strong>We developed an alignment-free framework that utilizes machine learning approaches for an ultra-fast and highly accurate prediction of the lethality of human-adapted coronavirus using genomic sequences. We performed extensive experiments through six different feature transformation and machine learning algorithms combining digital signal processing to identify the lethality of possible future novel coronaviruses using existing strains.</p><p><strong>Results: </strong>The results tested on SARS-CoV, MERS-CoV and SARS-CoV-2 datasets show an average 96.7% prediction accuracy. We also provide preliminary analysis validating the effectiveness of our models through other human coronaviruses. Our framework achieves high levels of prediction performance that is alignment-free and based on RNA sequences alone without genome annotations and specialized biological knowledge.</p><p><strong>Conclusion: </strong>The results demonstrate that, for any novel human coronavirus strains, this study can offer a reliable real-time estimation for its viral lethality.</p>","PeriodicalId":10803,"journal":{"name":"Current Genomics","volume":"22 1","pages":"583-595"},"PeriodicalIF":1.4000,"publicationDate":"2021-12-31","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8922323/pdf/","citationCount":"0","resultStr":"{\"title\":\"Exploring the Lethality of Human-Adapted Coronavirus Through Alignment-Free Machine Learning Approaches Using Genomic Sequences.\",\"authors\":\"Rui Yin, Zihan Luo, Chee Keong Kwoh\",\"doi\":\"10.2174/1389202923666211221110857\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>A newly emerging novel coronavirus appeared and rapidly spread worldwide and World Health Organization declared a pandemic on March 11, 2020. The roles and characteristics of coronavirus have captured much attention due to its power of causing a wide variety of infectious diseases, from mild to severe, on humans. The detection of the lethality of human coronavirus is key to estimate the viral toxicity and provide perspectives for treatment.</p><p><strong>Methods: </strong>We developed an alignment-free framework that utilizes machine learning approaches for an ultra-fast and highly accurate prediction of the lethality of human-adapted coronavirus using genomic sequences. We performed extensive experiments through six different feature transformation and machine learning algorithms combining digital signal processing to identify the lethality of possible future novel coronaviruses using existing strains.</p><p><strong>Results: </strong>The results tested on SARS-CoV, MERS-CoV and SARS-CoV-2 datasets show an average 96.7% prediction accuracy. We also provide preliminary analysis validating the effectiveness of our models through other human coronaviruses. Our framework achieves high levels of prediction performance that is alignment-free and based on RNA sequences alone without genome annotations and specialized biological knowledge.</p><p><strong>Conclusion: </strong>The results demonstrate that, for any novel human coronavirus strains, this study can offer a reliable real-time estimation for its viral lethality.</p>\",\"PeriodicalId\":10803,\"journal\":{\"name\":\"Current Genomics\",\"volume\":\"22 1\",\"pages\":\"583-595\"},\"PeriodicalIF\":1.4000,\"publicationDate\":\"2021-12-31\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8922323/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Current Genomics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.2174/1389202923666211221110857\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Current Genomics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.2174/1389202923666211221110857","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
Exploring the Lethality of Human-Adapted Coronavirus Through Alignment-Free Machine Learning Approaches Using Genomic Sequences.
Background: A newly emerging novel coronavirus appeared and rapidly spread worldwide and World Health Organization declared a pandemic on March 11, 2020. The roles and characteristics of coronavirus have captured much attention due to its power of causing a wide variety of infectious diseases, from mild to severe, on humans. The detection of the lethality of human coronavirus is key to estimate the viral toxicity and provide perspectives for treatment.
Methods: We developed an alignment-free framework that utilizes machine learning approaches for an ultra-fast and highly accurate prediction of the lethality of human-adapted coronavirus using genomic sequences. We performed extensive experiments through six different feature transformation and machine learning algorithms combining digital signal processing to identify the lethality of possible future novel coronaviruses using existing strains.
Results: The results tested on SARS-CoV, MERS-CoV and SARS-CoV-2 datasets show an average 96.7% prediction accuracy. We also provide preliminary analysis validating the effectiveness of our models through other human coronaviruses. Our framework achieves high levels of prediction performance that is alignment-free and based on RNA sequences alone without genome annotations and specialized biological knowledge.
Conclusion: The results demonstrate that, for any novel human coronavirus strains, this study can offer a reliable real-time estimation for its viral lethality.
期刊介绍:
Current Genomics is a peer-reviewed journal that provides essential reading about the latest and most important developments in genome science and related fields of research. Systems biology, systems modeling, machine learning, network inference, bioinformatics, computational biology, epigenetics, single cell genomics, extracellular vesicles, quantitative biology, and synthetic biology for the study of evolution, development, maintenance, aging and that of human health, human diseases, clinical genomics and precision medicine are topics of particular interest. The journal covers plant genomics. The journal will not consider articles dealing with breeding and livestock.
Current Genomics publishes three types of articles including:
i) Research papers from internationally-recognized experts reporting on new and original data generated at the genome scale level. Position papers dealing with new or challenging methodological approaches, whether experimental or mathematical, are greatly welcome in this section.
ii) Authoritative and comprehensive full-length or mini reviews from widely recognized experts, covering the latest developments in genome science and related fields of research such as systems biology, statistics and machine learning, quantitative biology, and precision medicine. Proposals for mini-hot topics (2-3 review papers) and full hot topics (6-8 review papers) guest edited by internationally-recognized experts are welcome in this section. Hot topic proposals should not contain original data and they should contain articles originating from at least 2 different countries.
iii) Opinion papers from internationally recognized experts addressing contemporary questions and issues in the field of genome science and systems biology and basic and clinical research practices.
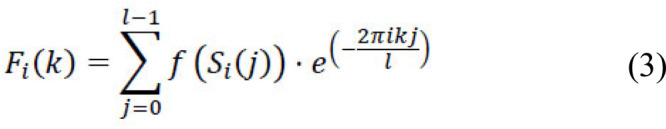
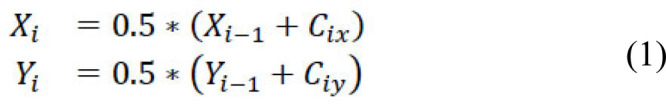
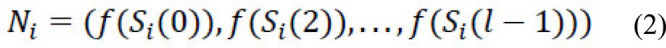

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


