{"title":"五名日本假软骨发育不全患者的新型和复发性COMP基因变异:从新生儿期到婴儿期的骨骼变化。","authors":"Kosei Hasegawa, Natsuko Futagawa, Yuko Ago, Hiroyuki Miyahara, Daisuke Harada, Mari Miyazawa, Junko Yoshimoto, Kenji Baba, Tadashi Moriwake, Hiroyuki Tanaka, Hirokazu Tsukahara","doi":"10.1297/cpe.2023-0035","DOIUrl":null,"url":null,"abstract":"<p><p>Pseudoachondroplasia (PSACH) is an autosomal dominant skeletal dysplasia caused by pathogenic variants of cartilage oligomeric matrix protein (COMP). Clinical symptoms of PSACH are characterized by growth disturbances after the first year of life. These disturbances lead to severe short stature with short limbs, brachydactyly, scoliosis, joint laxity, joint pain since childhood, and a normal face. Epimetaphyseal dysplasia, shortened long bones, and short metacarpals and phalanges are common findings on radiological examination. Additionally, anterior tonguing of the vertebral bodies in the lateral view is an important finding in childhood because it is specific to PSACH and normalizes with age. Here, we report five Japanese patients with PSACH, with one recurrent (p.Cys351Tyr) and four novel heterozygous pathogenic COMP variants (p.Asp437Tyr, p.Asp446Gly, p.Asp507Tyr, and p.Asp518Val). These five pathogenic variants were located in the calcium-binding type 3 (T3) repeats. In four of the novel variants, the affected amino acid was aspartic acid, which is abundant in each of the eight T3 repeats. We describe the radiological findings of these five patients. We also retrospectively analyzed the sequential changes in the vertebral body and epimetaphysis of the long bones from the neonatal to infantile periods in a patient with PSACH and congenital heart disease.</p>","PeriodicalId":10678,"journal":{"name":"Clinical Pediatric Endocrinology","volume":null,"pages":null},"PeriodicalIF":1.0000,"publicationDate":"2023-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/8c/d6/cpe-32-221.PMC10568574.pdf","citationCount":"0","resultStr":"{\"title\":\"Novel and recurrent <i>COMP</i> gene variants in five Japanese patients with pseudoachondroplasia: skeletal changes from the neonatal to infantile periods.\",\"authors\":\"Kosei Hasegawa, Natsuko Futagawa, Yuko Ago, Hiroyuki Miyahara, Daisuke Harada, Mari Miyazawa, Junko Yoshimoto, Kenji Baba, Tadashi Moriwake, Hiroyuki Tanaka, Hirokazu Tsukahara\",\"doi\":\"10.1297/cpe.2023-0035\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Pseudoachondroplasia (PSACH) is an autosomal dominant skeletal dysplasia caused by pathogenic variants of cartilage oligomeric matrix protein (COMP). Clinical symptoms of PSACH are characterized by growth disturbances after the first year of life. These disturbances lead to severe short stature with short limbs, brachydactyly, scoliosis, joint laxity, joint pain since childhood, and a normal face. Epimetaphyseal dysplasia, shortened long bones, and short metacarpals and phalanges are common findings on radiological examination. Additionally, anterior tonguing of the vertebral bodies in the lateral view is an important finding in childhood because it is specific to PSACH and normalizes with age. Here, we report five Japanese patients with PSACH, with one recurrent (p.Cys351Tyr) and four novel heterozygous pathogenic COMP variants (p.Asp437Tyr, p.Asp446Gly, p.Asp507Tyr, and p.Asp518Val). These five pathogenic variants were located in the calcium-binding type 3 (T3) repeats. In four of the novel variants, the affected amino acid was aspartic acid, which is abundant in each of the eight T3 repeats. We describe the radiological findings of these five patients. We also retrospectively analyzed the sequential changes in the vertebral body and epimetaphysis of the long bones from the neonatal to infantile periods in a patient with PSACH and congenital heart disease.</p>\",\"PeriodicalId\":10678,\"journal\":{\"name\":\"Clinical Pediatric Endocrinology\",\"volume\":null,\"pages\":null},\"PeriodicalIF\":1.0000,\"publicationDate\":\"2023-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/8c/d6/cpe-32-221.PMC10568574.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Clinical Pediatric Endocrinology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1297/cpe.2023-0035\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/9/16 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q4\",\"JCRName\":\"ENDOCRINOLOGY & METABOLISM\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical Pediatric Endocrinology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1297/cpe.2023-0035","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/9/16 0:00:00","PubModel":"Epub","JCR":"Q4","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
Novel and recurrent COMP gene variants in five Japanese patients with pseudoachondroplasia: skeletal changes from the neonatal to infantile periods.
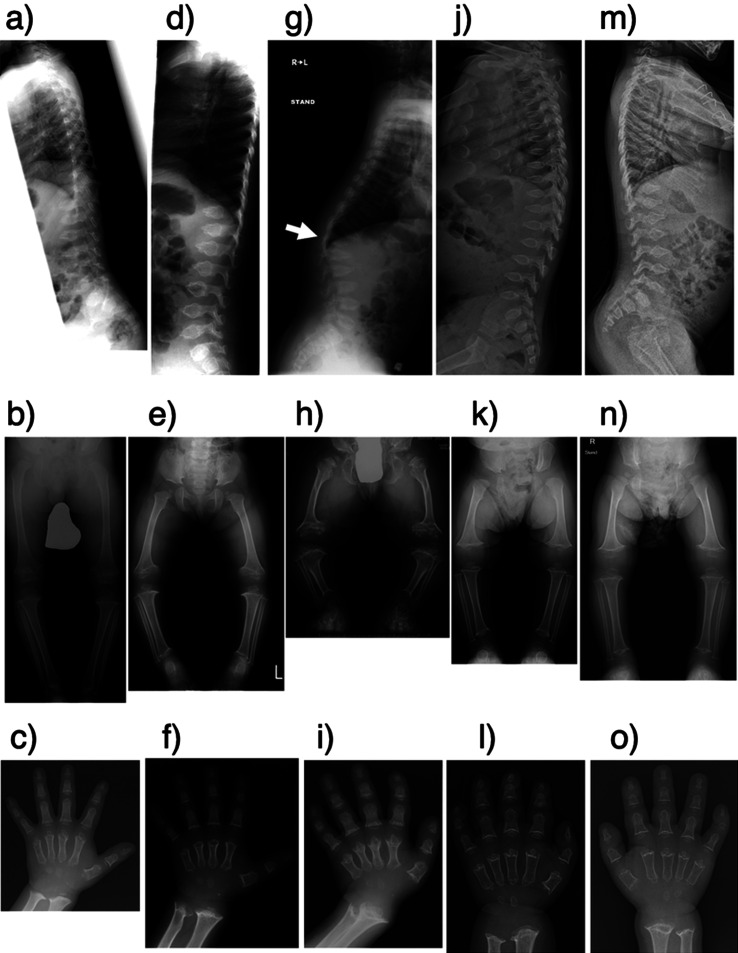
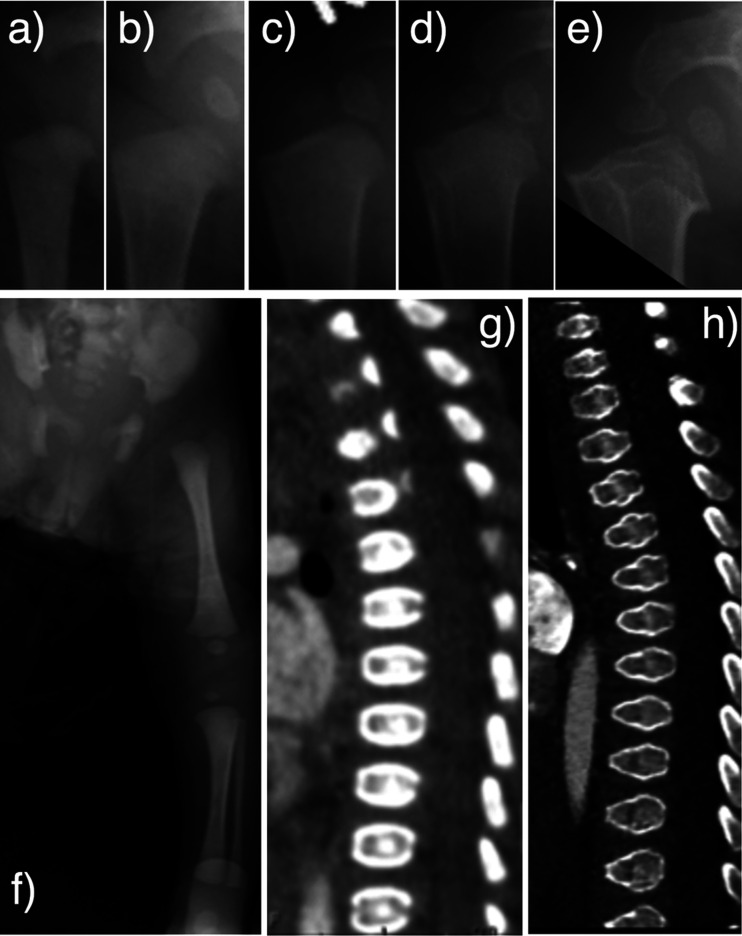
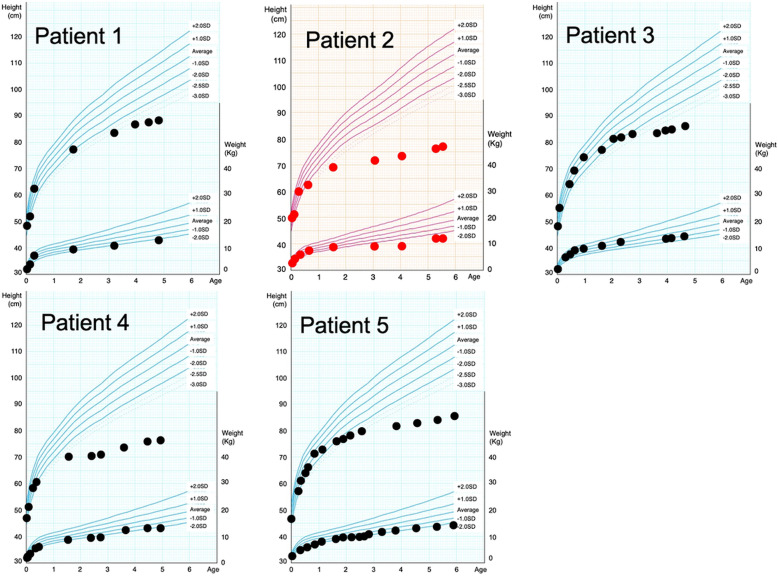
Pseudoachondroplasia (PSACH) is an autosomal dominant skeletal dysplasia caused by pathogenic variants of cartilage oligomeric matrix protein (COMP). Clinical symptoms of PSACH are characterized by growth disturbances after the first year of life. These disturbances lead to severe short stature with short limbs, brachydactyly, scoliosis, joint laxity, joint pain since childhood, and a normal face. Epimetaphyseal dysplasia, shortened long bones, and short metacarpals and phalanges are common findings on radiological examination. Additionally, anterior tonguing of the vertebral bodies in the lateral view is an important finding in childhood because it is specific to PSACH and normalizes with age. Here, we report five Japanese patients with PSACH, with one recurrent (p.Cys351Tyr) and four novel heterozygous pathogenic COMP variants (p.Asp437Tyr, p.Asp446Gly, p.Asp507Tyr, and p.Asp518Val). These five pathogenic variants were located in the calcium-binding type 3 (T3) repeats. In four of the novel variants, the affected amino acid was aspartic acid, which is abundant in each of the eight T3 repeats. We describe the radiological findings of these five patients. We also retrospectively analyzed the sequential changes in the vertebral body and epimetaphysis of the long bones from the neonatal to infantile periods in a patient with PSACH and congenital heart disease.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


