Ben J Hayes, James Copley, Elsie Dodd, Elizabeth M Ross, Shannon Speight, Geoffry Fordyce
{"title":"在没有谱系信息的情况下对热带肉牛进行多品种基因组评估。","authors":"Ben J Hayes, James Copley, Elsie Dodd, Elizabeth M Ross, Shannon Speight, Geoffry Fordyce","doi":"10.1186/s12711-023-00847-6","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>It has been challenging to implement genomic selection in multi-breed tropical beef cattle populations. If commercial (often crossbred) animals could be used in the reference population for these genomic evaluations, this could allow for very large reference populations. In tropical beef systems, such animals often have no pedigree information. Here we investigate potential models for such data, using marker heterozygosity (to model heterosis) and breed composition derived from genetic markers, as covariates in the model. Models treated breed effects as either fixed or random, and included genomic best linear unbiased prediction (GBLUP) and BayesR. A tropically-adapted beef cattle dataset of 29,391 purebred, crossbred and composite commercial animals was used to evaluate the models.</p><p><strong>Results: </strong>Treating breed effects as random, in an approach analogous to genetic groups allowed partitioning of the genetic variance into within-breed and across breed-components (even with a large number of breeds), and estimation of within-breed and across-breed genomic estimated breeding values (GEBV). We demonstrate that moderately-accurate (0.30-0.43) GEBV can be calculated using these models. Treating breed effects as random gave more accurate GEBV than treating breed as fixed. A simple GBLUP model where no breed effects were fitted gave the same accuracy (and correlations of GEBV very close to 1) as a model where GEBV for within-breed and the GEBV for (random) across-breed effects were included. When GEBV were predicted for herds with no data in the reference population, BayesR resulted in the highest accuracy, with 3% accuracy improvement averaged across traits, especially when the validation population was less related to the reference population. Estimates of heterosis from our models were in line with previous estimates from beef cattle. A method for estimating the number of effective breed comparisons for each breed combination accumulated across contemporary groups is presented.</p><p><strong>Conclusions: </strong>When no pedigree is available, breed composition and heterosis for inclusion in multi-breed genomic evaluation can be estimated from genotypes. When GEBV were predicted for herds with no data in the reference population, BayesR resulted in the highest accuracy.</p>","PeriodicalId":55120,"journal":{"name":"Genetics Selection Evolution","volume":"55 1","pages":"71"},"PeriodicalIF":3.1000,"publicationDate":"2023-10-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10578004/pdf/","citationCount":"1","resultStr":"{\"title\":\"Multi-breed genomic evaluation for tropical beef cattle when no pedigree information is available.\",\"authors\":\"Ben J Hayes, James Copley, Elsie Dodd, Elizabeth M Ross, Shannon Speight, Geoffry Fordyce\",\"doi\":\"10.1186/s12711-023-00847-6\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>It has been challenging to implement genomic selection in multi-breed tropical beef cattle populations. If commercial (often crossbred) animals could be used in the reference population for these genomic evaluations, this could allow for very large reference populations. In tropical beef systems, such animals often have no pedigree information. Here we investigate potential models for such data, using marker heterozygosity (to model heterosis) and breed composition derived from genetic markers, as covariates in the model. Models treated breed effects as either fixed or random, and included genomic best linear unbiased prediction (GBLUP) and BayesR. A tropically-adapted beef cattle dataset of 29,391 purebred, crossbred and composite commercial animals was used to evaluate the models.</p><p><strong>Results: </strong>Treating breed effects as random, in an approach analogous to genetic groups allowed partitioning of the genetic variance into within-breed and across breed-components (even with a large number of breeds), and estimation of within-breed and across-breed genomic estimated breeding values (GEBV). We demonstrate that moderately-accurate (0.30-0.43) GEBV can be calculated using these models. Treating breed effects as random gave more accurate GEBV than treating breed as fixed. A simple GBLUP model where no breed effects were fitted gave the same accuracy (and correlations of GEBV very close to 1) as a model where GEBV for within-breed and the GEBV for (random) across-breed effects were included. When GEBV were predicted for herds with no data in the reference population, BayesR resulted in the highest accuracy, with 3% accuracy improvement averaged across traits, especially when the validation population was less related to the reference population. Estimates of heterosis from our models were in line with previous estimates from beef cattle. A method for estimating the number of effective breed comparisons for each breed combination accumulated across contemporary groups is presented.</p><p><strong>Conclusions: </strong>When no pedigree is available, breed composition and heterosis for inclusion in multi-breed genomic evaluation can be estimated from genotypes. When GEBV were predicted for herds with no data in the reference population, BayesR resulted in the highest accuracy.</p>\",\"PeriodicalId\":55120,\"journal\":{\"name\":\"Genetics Selection Evolution\",\"volume\":\"55 1\",\"pages\":\"71\"},\"PeriodicalIF\":3.1000,\"publicationDate\":\"2023-10-16\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10578004/pdf/\",\"citationCount\":\"1\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Genetics Selection Evolution\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1186/s12711-023-00847-6\",\"RegionNum\":1,\"RegionCategory\":\"农林科学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"AGRICULTURE, DAIRY & ANIMAL SCIENCE\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Genetics Selection Evolution","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s12711-023-00847-6","RegionNum":1,"RegionCategory":"农林科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"AGRICULTURE, DAIRY & ANIMAL SCIENCE","Score":null,"Total":0}
Multi-breed genomic evaluation for tropical beef cattle when no pedigree information is available.
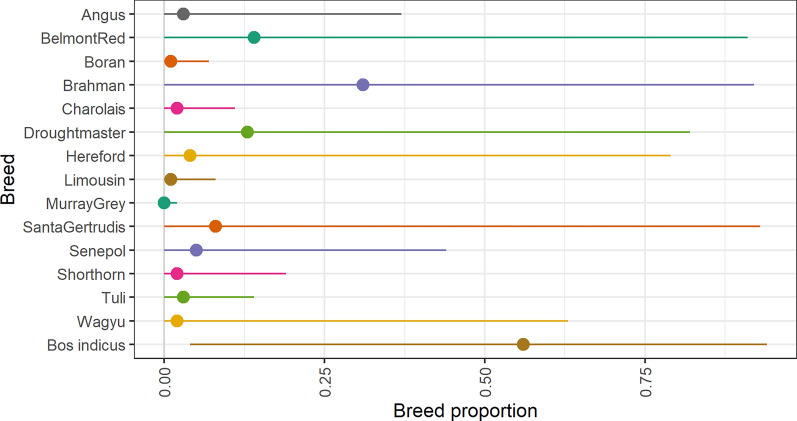
Background: It has been challenging to implement genomic selection in multi-breed tropical beef cattle populations. If commercial (often crossbred) animals could be used in the reference population for these genomic evaluations, this could allow for very large reference populations. In tropical beef systems, such animals often have no pedigree information. Here we investigate potential models for such data, using marker heterozygosity (to model heterosis) and breed composition derived from genetic markers, as covariates in the model. Models treated breed effects as either fixed or random, and included genomic best linear unbiased prediction (GBLUP) and BayesR. A tropically-adapted beef cattle dataset of 29,391 purebred, crossbred and composite commercial animals was used to evaluate the models.
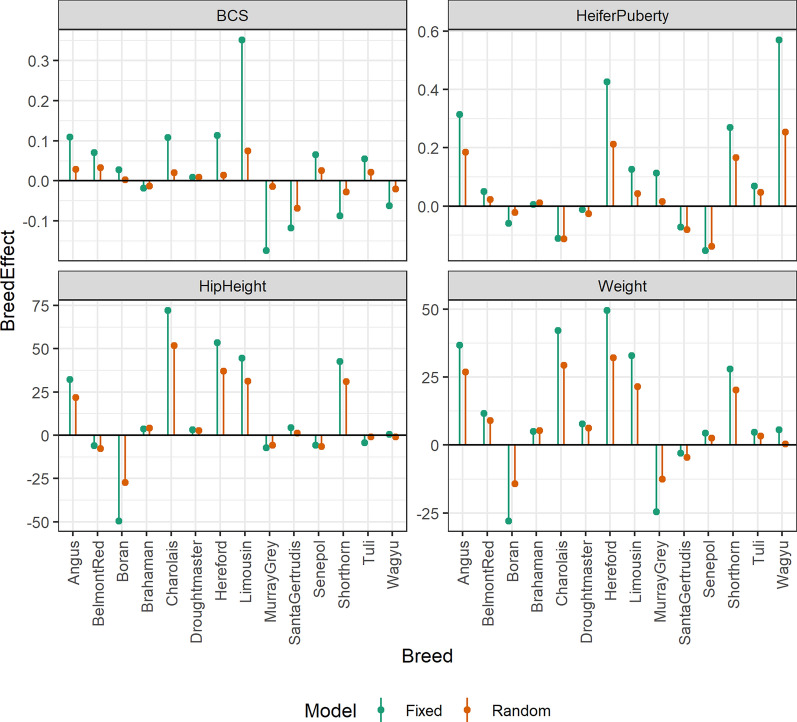
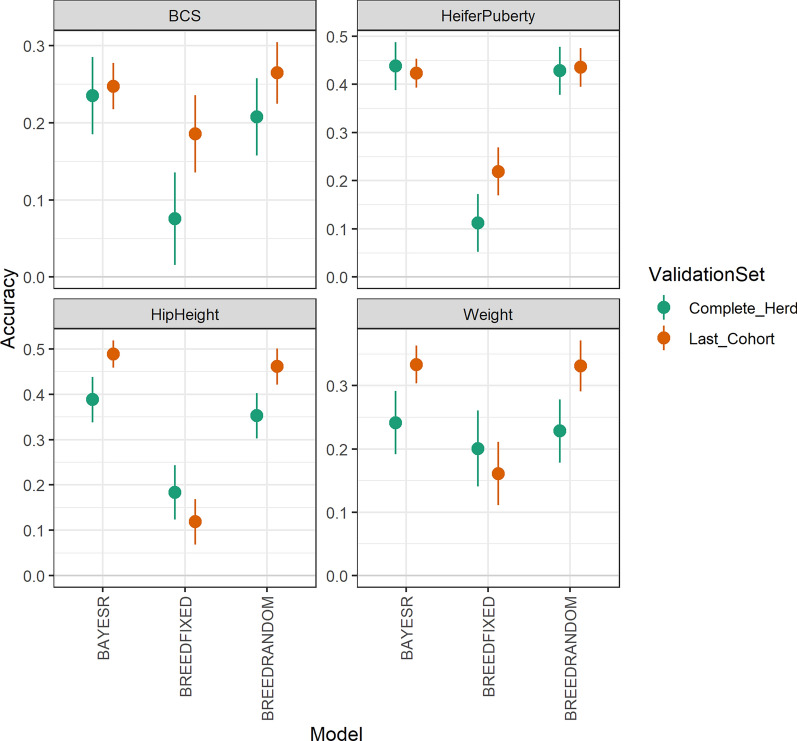
Results: Treating breed effects as random, in an approach analogous to genetic groups allowed partitioning of the genetic variance into within-breed and across breed-components (even with a large number of breeds), and estimation of within-breed and across-breed genomic estimated breeding values (GEBV). We demonstrate that moderately-accurate (0.30-0.43) GEBV can be calculated using these models. Treating breed effects as random gave more accurate GEBV than treating breed as fixed. A simple GBLUP model where no breed effects were fitted gave the same accuracy (and correlations of GEBV very close to 1) as a model where GEBV for within-breed and the GEBV for (random) across-breed effects were included. When GEBV were predicted for herds with no data in the reference population, BayesR resulted in the highest accuracy, with 3% accuracy improvement averaged across traits, especially when the validation population was less related to the reference population. Estimates of heterosis from our models were in line with previous estimates from beef cattle. A method for estimating the number of effective breed comparisons for each breed combination accumulated across contemporary groups is presented.
Conclusions: When no pedigree is available, breed composition and heterosis for inclusion in multi-breed genomic evaluation can be estimated from genotypes. When GEBV were predicted for herds with no data in the reference population, BayesR resulted in the highest accuracy.
期刊介绍:
Genetics Selection Evolution invites basic, applied and methodological content that will aid the current understanding and the utilization of genetic variability in domestic animal species. Although the focus is on domestic animal species, research on other species is invited if it contributes to the understanding of the use of genetic variability in domestic animals. Genetics Selection Evolution publishes results from all levels of study, from the gene to the quantitative trait, from the individual to the population, the breed or the species. Contributions concerning both the biological approach, from molecular genetics to quantitative genetics, as well as the mathematical approach, from population genetics to statistics, are welcome. Specific areas of interest include but are not limited to: gene and QTL identification, mapping and characterization, analysis of new phenotypes, high-throughput SNP data analysis, functional genomics, cytogenetics, genetic diversity of populations and breeds, genetic evaluation, applied and experimental selection, genomic selection, selection efficiency, and statistical methodology for the genetic analysis of phenotypes with quantitative and mixed inheritance.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


