Daniela Felício, Tanguy Rubat du Mérac, António Amorim, Sandra Martins
{"title":"多谷氨酰胺脊髓小脑共济失调中旁系基因的功能意义。","authors":"Daniela Felício, Tanguy Rubat du Mérac, António Amorim, Sandra Martins","doi":"10.1007/s00439-023-02607-4","DOIUrl":null,"url":null,"abstract":"<p><p>Polyglutamine (polyQ) spinocerebellar ataxias (SCAs) comprise a group of autosomal dominant neurodegenerative disorders caused by (CAG/CAA)<sub>n</sub> expansions. The elongated stretches of adjacent glutamines alter the conformation of the native proteins inducing neurotoxicity, and subsequent motor and neurological symptoms. Although the etiology and neuropathology of most polyQ SCAs have been extensively studied, only a limited selection of therapies is available. Previous studies on SCA1 demonstrated that ATXN1L, a human duplicated gene of the disease-associated ATXN1, alleviated neuropathology in mice models. Other SCA-associated genes have paralogs (i.e., copies at different chromosomal locations derived from duplication of the parental gene), but their functional relevance and potential role in disease pathogenesis remain unexplored. Here, we review the protein homology, expression pattern, and molecular functions of paralogs in seven polyQ dominant ataxias-SCA1, SCA2, MJD/SCA3, SCA6, SCA7, SCA17, and DRPLA. Besides ATXN1L, we highlight ATXN2L, ATXN3L, CACNA1B, ATXN7L1, ATXN7L2, TBPL2, and RERE as promising functional candidates to play a role in the neuropathology of the respective SCA, along with the parental gene. Although most of these duplicates lack the (CAG/CAA)<sub>n</sub> region, if functionally redundant, they may compensate for a partial loss-of-function or dysfunction of the wild-type genes in SCAs. We aim to draw attention to the hypothesis that paralogs of disease-associated genes may underlie the complex neuropathology of dominant ataxias and potentiate new therapeutic strategies.</p>","PeriodicalId":13175,"journal":{"name":"Human Genetics","volume":null,"pages":null},"PeriodicalIF":3.8000,"publicationDate":"2023-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10676324/pdf/","citationCount":"0","resultStr":"{\"title\":\"Functional implications of paralog genes in polyglutamine spinocerebellar ataxias.\",\"authors\":\"Daniela Felício, Tanguy Rubat du Mérac, António Amorim, Sandra Martins\",\"doi\":\"10.1007/s00439-023-02607-4\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Polyglutamine (polyQ) spinocerebellar ataxias (SCAs) comprise a group of autosomal dominant neurodegenerative disorders caused by (CAG/CAA)<sub>n</sub> expansions. The elongated stretches of adjacent glutamines alter the conformation of the native proteins inducing neurotoxicity, and subsequent motor and neurological symptoms. Although the etiology and neuropathology of most polyQ SCAs have been extensively studied, only a limited selection of therapies is available. Previous studies on SCA1 demonstrated that ATXN1L, a human duplicated gene of the disease-associated ATXN1, alleviated neuropathology in mice models. Other SCA-associated genes have paralogs (i.e., copies at different chromosomal locations derived from duplication of the parental gene), but their functional relevance and potential role in disease pathogenesis remain unexplored. Here, we review the protein homology, expression pattern, and molecular functions of paralogs in seven polyQ dominant ataxias-SCA1, SCA2, MJD/SCA3, SCA6, SCA7, SCA17, and DRPLA. Besides ATXN1L, we highlight ATXN2L, ATXN3L, CACNA1B, ATXN7L1, ATXN7L2, TBPL2, and RERE as promising functional candidates to play a role in the neuropathology of the respective SCA, along with the parental gene. Although most of these duplicates lack the (CAG/CAA)<sub>n</sub> region, if functionally redundant, they may compensate for a partial loss-of-function or dysfunction of the wild-type genes in SCAs. We aim to draw attention to the hypothesis that paralogs of disease-associated genes may underlie the complex neuropathology of dominant ataxias and potentiate new therapeutic strategies.</p>\",\"PeriodicalId\":13175,\"journal\":{\"name\":\"Human Genetics\",\"volume\":null,\"pages\":null},\"PeriodicalIF\":3.8000,\"publicationDate\":\"2023-12-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10676324/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Human Genetics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1007/s00439-023-02607-4\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/10/16 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Human Genetics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1007/s00439-023-02607-4","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/10/16 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Functional implications of paralog genes in polyglutamine spinocerebellar ataxias.
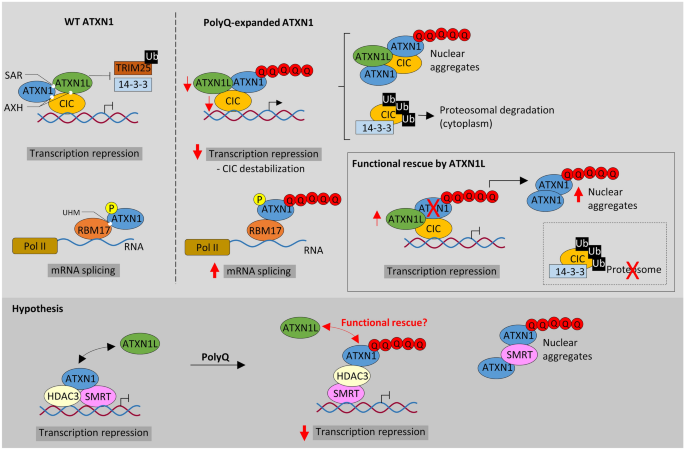
Polyglutamine (polyQ) spinocerebellar ataxias (SCAs) comprise a group of autosomal dominant neurodegenerative disorders caused by (CAG/CAA)n expansions. The elongated stretches of adjacent glutamines alter the conformation of the native proteins inducing neurotoxicity, and subsequent motor and neurological symptoms. Although the etiology and neuropathology of most polyQ SCAs have been extensively studied, only a limited selection of therapies is available. Previous studies on SCA1 demonstrated that ATXN1L, a human duplicated gene of the disease-associated ATXN1, alleviated neuropathology in mice models. Other SCA-associated genes have paralogs (i.e., copies at different chromosomal locations derived from duplication of the parental gene), but their functional relevance and potential role in disease pathogenesis remain unexplored. Here, we review the protein homology, expression pattern, and molecular functions of paralogs in seven polyQ dominant ataxias-SCA1, SCA2, MJD/SCA3, SCA6, SCA7, SCA17, and DRPLA. Besides ATXN1L, we highlight ATXN2L, ATXN3L, CACNA1B, ATXN7L1, ATXN7L2, TBPL2, and RERE as promising functional candidates to play a role in the neuropathology of the respective SCA, along with the parental gene. Although most of these duplicates lack the (CAG/CAA)n region, if functionally redundant, they may compensate for a partial loss-of-function or dysfunction of the wild-type genes in SCAs. We aim to draw attention to the hypothesis that paralogs of disease-associated genes may underlie the complex neuropathology of dominant ataxias and potentiate new therapeutic strategies.
期刊介绍:
Human Genetics is a monthly journal publishing original and timely articles on all aspects of human genetics. The Journal particularly welcomes articles in the areas of Behavioral genetics, Bioinformatics, Cancer genetics and genomics, Cytogenetics, Developmental genetics, Disease association studies, Dysmorphology, ELSI (ethical, legal and social issues), Evolutionary genetics, Gene expression, Gene structure and organization, Genetics of complex diseases and epistatic interactions, Genetic epidemiology, Genome biology, Genome structure and organization, Genotype-phenotype relationships, Human Genomics, Immunogenetics and genomics, Linkage analysis and genetic mapping, Methods in Statistical Genetics, Molecular diagnostics, Mutation detection and analysis, Neurogenetics, Physical mapping and Population Genetics. Articles reporting animal models relevant to human biology or disease are also welcome. Preference will be given to those articles which address clinically relevant questions or which provide new insights into human biology.
Unless reporting entirely novel and unusual aspects of a topic, clinical case reports, cytogenetic case reports, papers on descriptive population genetics, articles dealing with the frequency of polymorphisms or additional mutations within genes in which numerous lesions have already been described, and papers that report meta-analyses of previously published datasets will normally not be accepted.
The Journal typically will not consider for publication manuscripts that report merely the isolation, map position, structure, and tissue expression profile of a gene of unknown function unless the gene is of particular interest or is a candidate gene involved in a human trait or disorder.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


