Juan V. Alegre-Requena, Shree Sowndarya S. V., Raúl Pérez-Soto, Turki M. Alturaifi, Robert S. Paton
{"title":"AQME:研究人员和教育工作者的自动化量子力学环境","authors":"Juan V. Alegre-Requena, Shree Sowndarya S. V., Raúl Pérez-Soto, Turki M. Alturaifi, Robert S. Paton","doi":"10.1002/wcms.1663","DOIUrl":null,"url":null,"abstract":"<p>AQME, automated quantum mechanical environments, is a free and open-source Python package for the rapid deployment of automated workflows using cheminformatics and quantum chemistry. AQME workflows integrate tasks performed across multiple computational chemistry packages and data formats, preserving all computational protocols, data, and metadata for machine and human users to access and reuse. AQME has a modular structure of independent modules that can be implemented in any sequence, allowing the users to use all or only the desired parts of the program. The code has been developed for researchers with basic familiarity with the Python programming language. The CSEARCH module interfaces to molecular mechanics and semi-empirical QM (SQM) conformer generation tools (e.g., RDKit and Conformer–Rotamer Ensemble Sampling Tool, CREST) starting from various initial structure formats. The CMIN module enables geometry refinement with SQM and neural network potentials, such as ANI. The QPREP module interfaces with multiple QM programs, such as Gaussian, ORCA, and PySCF. The QCORR module processes QM results, storing structural, energetic, and property data while also enabling automated error handling (i.e., convergence errors, wrong number of imaginary frequencies, isomerization, etc.) and job resubmission. The QDESCP module provides easy access to QM ensemble-averaged molecular descriptors and computed properties, such as NMR spectra. Overall, AQME provides automated, transparent, and reproducible workflows to produce, analyze and archive computational chemistry results. SMILES inputs can be used, and many aspects of tedious human manipulation can be avoided. Installation and execution on Windows, macOS, and Linux platforms have been tested, and the code has been developed to support access through Jupyter Notebooks, the command line, and job submission (e.g., Slurm) scripts. Examples of pre-configured workflows are available in various formats, and hands-on video tutorials illustrate their use.</p><p>This article is categorized under:\n </p>","PeriodicalId":236,"journal":{"name":"Wiley Interdisciplinary Reviews: Computational Molecular Science","volume":"13 5","pages":""},"PeriodicalIF":16.8000,"publicationDate":"2023-02-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/wcms.1663","citationCount":"3","resultStr":"{\"title\":\"AQME: Automated quantum mechanical environments for researchers and educators\",\"authors\":\"Juan V. Alegre-Requena, Shree Sowndarya S. V., Raúl Pérez-Soto, Turki M. Alturaifi, Robert S. Paton\",\"doi\":\"10.1002/wcms.1663\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>AQME, automated quantum mechanical environments, is a free and open-source Python package for the rapid deployment of automated workflows using cheminformatics and quantum chemistry. AQME workflows integrate tasks performed across multiple computational chemistry packages and data formats, preserving all computational protocols, data, and metadata for machine and human users to access and reuse. AQME has a modular structure of independent modules that can be implemented in any sequence, allowing the users to use all or only the desired parts of the program. The code has been developed for researchers with basic familiarity with the Python programming language. The CSEARCH module interfaces to molecular mechanics and semi-empirical QM (SQM) conformer generation tools (e.g., RDKit and Conformer–Rotamer Ensemble Sampling Tool, CREST) starting from various initial structure formats. The CMIN module enables geometry refinement with SQM and neural network potentials, such as ANI. The QPREP module interfaces with multiple QM programs, such as Gaussian, ORCA, and PySCF. The QCORR module processes QM results, storing structural, energetic, and property data while also enabling automated error handling (i.e., convergence errors, wrong number of imaginary frequencies, isomerization, etc.) and job resubmission. The QDESCP module provides easy access to QM ensemble-averaged molecular descriptors and computed properties, such as NMR spectra. Overall, AQME provides automated, transparent, and reproducible workflows to produce, analyze and archive computational chemistry results. SMILES inputs can be used, and many aspects of tedious human manipulation can be avoided. Installation and execution on Windows, macOS, and Linux platforms have been tested, and the code has been developed to support access through Jupyter Notebooks, the command line, and job submission (e.g., Slurm) scripts. Examples of pre-configured workflows are available in various formats, and hands-on video tutorials illustrate their use.</p><p>This article is categorized under:\\n </p>\",\"PeriodicalId\":236,\"journal\":{\"name\":\"Wiley Interdisciplinary Reviews: Computational Molecular Science\",\"volume\":\"13 5\",\"pages\":\"\"},\"PeriodicalIF\":16.8000,\"publicationDate\":\"2023-02-26\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/wcms.1663\",\"citationCount\":\"3\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Wiley Interdisciplinary Reviews: Computational Molecular Science\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/wcms.1663\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Wiley Interdisciplinary Reviews: Computational Molecular Science","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/wcms.1663","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
AQME: Automated quantum mechanical environments for researchers and educators
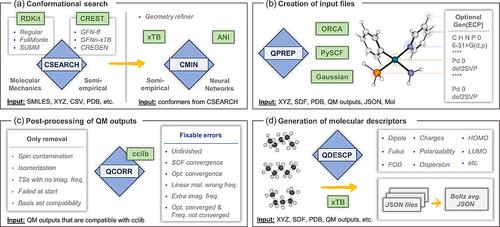
AQME, automated quantum mechanical environments, is a free and open-source Python package for the rapid deployment of automated workflows using cheminformatics and quantum chemistry. AQME workflows integrate tasks performed across multiple computational chemistry packages and data formats, preserving all computational protocols, data, and metadata for machine and human users to access and reuse. AQME has a modular structure of independent modules that can be implemented in any sequence, allowing the users to use all or only the desired parts of the program. The code has been developed for researchers with basic familiarity with the Python programming language. The CSEARCH module interfaces to molecular mechanics and semi-empirical QM (SQM) conformer generation tools (e.g., RDKit and Conformer–Rotamer Ensemble Sampling Tool, CREST) starting from various initial structure formats. The CMIN module enables geometry refinement with SQM and neural network potentials, such as ANI. The QPREP module interfaces with multiple QM programs, such as Gaussian, ORCA, and PySCF. The QCORR module processes QM results, storing structural, energetic, and property data while also enabling automated error handling (i.e., convergence errors, wrong number of imaginary frequencies, isomerization, etc.) and job resubmission. The QDESCP module provides easy access to QM ensemble-averaged molecular descriptors and computed properties, such as NMR spectra. Overall, AQME provides automated, transparent, and reproducible workflows to produce, analyze and archive computational chemistry results. SMILES inputs can be used, and many aspects of tedious human manipulation can be avoided. Installation and execution on Windows, macOS, and Linux platforms have been tested, and the code has been developed to support access through Jupyter Notebooks, the command line, and job submission (e.g., Slurm) scripts. Examples of pre-configured workflows are available in various formats, and hands-on video tutorials illustrate their use.
期刊介绍:
Computational molecular sciences harness the power of rigorous chemical and physical theories, employing computer-based modeling, specialized hardware, software development, algorithm design, and database management to explore and illuminate every facet of molecular sciences. These interdisciplinary approaches form a bridge between chemistry, biology, and materials sciences, establishing connections with adjacent application-driven fields in both chemistry and biology. WIREs Computational Molecular Science stands as a platform to comprehensively review and spotlight research from these dynamic and interconnected fields.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


