Francesco Di Palma, Carlo Abate, Sergio Decherchi, Andrea Cavalli
{"title":"通过机器学习进行可连接性和可药用性评估","authors":"Francesco Di Palma, Carlo Abate, Sergio Decherchi, Andrea Cavalli","doi":"10.1002/wcms.1676","DOIUrl":null,"url":null,"abstract":"<p>Drug discovery is a daunting and failure-prone task. A critical process in this research field is represented by the biological target and pocket identification steps as they heavily determine the subsequent efforts in selecting a putative ligand, most often a small molecule. Finding “ligandable” pockets, namely protein cavities that may accept a drug-like binder is instrumental to the more general and drug discovery oriented “druggability” estimation process. While high-throughput experimental techniques exist to identify putative binding sites other than the orthosteric one, these techniques are relatively expensive and not so commonly available in labs. In this regard, computational means of detecting ligandable pockets are advisable for their inexpensiveness and speed. These methods can become, in principle, particularly predictive when supported by machine learning methodologies that provide the modeling framework. As with any data-driven effort, the outcome critically depends on the input data, its featurization process and possible associated biases. Also, the machine learning task, (supervised/unsupervised) the learning method, and the possible usage of molecular dynamics data considerably shape the inherent assumptions of the modeling step. Defining a proper quantitative thermodynamic and/or kinetic score (or label) is key to the modeling process; here we revise literature and propose residence time as a novel ideal indicator of ligandability. Interestingly the vast majority of the methods does not keep into consideration kinetics nor thermodynamics when devising predictors.</p><p>This article is categorized under:\n </p>","PeriodicalId":236,"journal":{"name":"Wiley Interdisciplinary Reviews: Computational Molecular Science","volume":"13 5","pages":""},"PeriodicalIF":27.0000,"publicationDate":"2023-06-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/wcms.1676","citationCount":"1","resultStr":"{\"title\":\"Ligandability and druggability assessment via machine learning\",\"authors\":\"Francesco Di Palma, Carlo Abate, Sergio Decherchi, Andrea Cavalli\",\"doi\":\"10.1002/wcms.1676\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Drug discovery is a daunting and failure-prone task. A critical process in this research field is represented by the biological target and pocket identification steps as they heavily determine the subsequent efforts in selecting a putative ligand, most often a small molecule. Finding “ligandable” pockets, namely protein cavities that may accept a drug-like binder is instrumental to the more general and drug discovery oriented “druggability” estimation process. While high-throughput experimental techniques exist to identify putative binding sites other than the orthosteric one, these techniques are relatively expensive and not so commonly available in labs. In this regard, computational means of detecting ligandable pockets are advisable for their inexpensiveness and speed. These methods can become, in principle, particularly predictive when supported by machine learning methodologies that provide the modeling framework. As with any data-driven effort, the outcome critically depends on the input data, its featurization process and possible associated biases. Also, the machine learning task, (supervised/unsupervised) the learning method, and the possible usage of molecular dynamics data considerably shape the inherent assumptions of the modeling step. Defining a proper quantitative thermodynamic and/or kinetic score (or label) is key to the modeling process; here we revise literature and propose residence time as a novel ideal indicator of ligandability. Interestingly the vast majority of the methods does not keep into consideration kinetics nor thermodynamics when devising predictors.</p><p>This article is categorized under:\\n </p>\",\"PeriodicalId\":236,\"journal\":{\"name\":\"Wiley Interdisciplinary Reviews: Computational Molecular Science\",\"volume\":\"13 5\",\"pages\":\"\"},\"PeriodicalIF\":27.0000,\"publicationDate\":\"2023-06-04\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/wcms.1676\",\"citationCount\":\"1\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Wiley Interdisciplinary Reviews: Computational Molecular Science\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/wcms.1676\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Wiley Interdisciplinary Reviews: Computational Molecular Science","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/wcms.1676","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
Ligandability and druggability assessment via machine learning
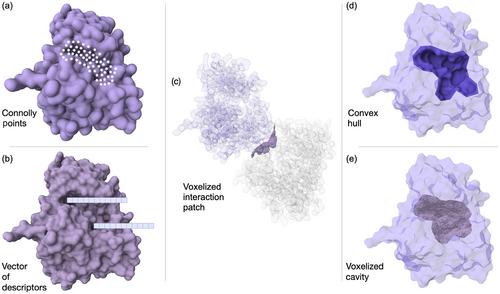
Drug discovery is a daunting and failure-prone task. A critical process in this research field is represented by the biological target and pocket identification steps as they heavily determine the subsequent efforts in selecting a putative ligand, most often a small molecule. Finding “ligandable” pockets, namely protein cavities that may accept a drug-like binder is instrumental to the more general and drug discovery oriented “druggability” estimation process. While high-throughput experimental techniques exist to identify putative binding sites other than the orthosteric one, these techniques are relatively expensive and not so commonly available in labs. In this regard, computational means of detecting ligandable pockets are advisable for their inexpensiveness and speed. These methods can become, in principle, particularly predictive when supported by machine learning methodologies that provide the modeling framework. As with any data-driven effort, the outcome critically depends on the input data, its featurization process and possible associated biases. Also, the machine learning task, (supervised/unsupervised) the learning method, and the possible usage of molecular dynamics data considerably shape the inherent assumptions of the modeling step. Defining a proper quantitative thermodynamic and/or kinetic score (or label) is key to the modeling process; here we revise literature and propose residence time as a novel ideal indicator of ligandability. Interestingly the vast majority of the methods does not keep into consideration kinetics nor thermodynamics when devising predictors.
期刊介绍:
Computational molecular sciences harness the power of rigorous chemical and physical theories, employing computer-based modeling, specialized hardware, software development, algorithm design, and database management to explore and illuminate every facet of molecular sciences. These interdisciplinary approaches form a bridge between chemistry, biology, and materials sciences, establishing connections with adjacent application-driven fields in both chemistry and biology. WIREs Computational Molecular Science stands as a platform to comprehensively review and spotlight research from these dynamic and interconnected fields.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


