{"title":"DynaDom:基于结构的T细胞受体结构域间和T细胞受体肽- mhc (I类)关联角预测","authors":"Thomas Hoffmann, Antoine Marion, Iris Antes","doi":"10.1186/s12900-016-0071-7","DOIUrl":null,"url":null,"abstract":"<p>T cell receptor (TCR) molecules are involved in the adaptive immune response as they distinguish between self- and foreign-peptides, presented in major histocompatibility complex molecules (pMHC). Former studies showed that the association angles of the TCR variable domains (Vα/Vβ) can differ significantly and change upon binding to the pMHC complex. These changes can be described as a rotation of the domains around a general Center of Rotation, characterized by the interaction of two highly conserved glutamine residues.</p><p>We developed a computational method, DynaDom, for the prediction of TCR Vα/Vβ inter-domain and TCR/pMHC orientations in TCRpMHC complexes, which allows predicting the orientation of multiple protein-domains. In addition, we implemented a new approach to predict the correct orientation of the carboxamide endgroups in glutamine and asparagine residues, which can also be used as an external, independent tool.</p><p>The approach was evaluated for the remodeling of 75 and 53 experimental structures of TCR and TCRpMHC (class I) complexes, respectively. We show that the DynaDom method predicts the correct orientation of the TCR Vα/Vβ angles in 96 and 89% of the cases, for the poses with the best RMSD and best interaction energy, respectively. For the concurrent prediction of the TCR Vα/Vβ and pMHC orientations, the respective rates reached 74 and 72%. Through an exhaustive analysis, we could show that the pMHC placement can be further improved by a straightforward, yet very time intensive extension of the current approach.</p><p>The results obtained in the present remodeling study prove the suitability of our approach for interdomain-angle optimization. In addition, the high prediction rate obtained specifically for the energetically highest ranked poses further demonstrates that our method is a powerful candidate for blind prediction. Therefore it should be well suited as part of any accurate atomistic modeling pipeline for TCRpMHC complexes and potentially other large molecular assemblies.</p>","PeriodicalId":498,"journal":{"name":"BMC Structural Biology","volume":"17 1","pages":""},"PeriodicalIF":2.2220,"publicationDate":"2017-02-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s12900-016-0071-7","citationCount":"11","resultStr":"{\"title\":\"DynaDom: structure-based prediction of T cell receptor inter-domain and T cell receptor-peptide-MHC (class I) association angles\",\"authors\":\"Thomas Hoffmann, Antoine Marion, Iris Antes\",\"doi\":\"10.1186/s12900-016-0071-7\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>T cell receptor (TCR) molecules are involved in the adaptive immune response as they distinguish between self- and foreign-peptides, presented in major histocompatibility complex molecules (pMHC). Former studies showed that the association angles of the TCR variable domains (Vα/Vβ) can differ significantly and change upon binding to the pMHC complex. These changes can be described as a rotation of the domains around a general Center of Rotation, characterized by the interaction of two highly conserved glutamine residues.</p><p>We developed a computational method, DynaDom, for the prediction of TCR Vα/Vβ inter-domain and TCR/pMHC orientations in TCRpMHC complexes, which allows predicting the orientation of multiple protein-domains. In addition, we implemented a new approach to predict the correct orientation of the carboxamide endgroups in glutamine and asparagine residues, which can also be used as an external, independent tool.</p><p>The approach was evaluated for the remodeling of 75 and 53 experimental structures of TCR and TCRpMHC (class I) complexes, respectively. We show that the DynaDom method predicts the correct orientation of the TCR Vα/Vβ angles in 96 and 89% of the cases, for the poses with the best RMSD and best interaction energy, respectively. For the concurrent prediction of the TCR Vα/Vβ and pMHC orientations, the respective rates reached 74 and 72%. Through an exhaustive analysis, we could show that the pMHC placement can be further improved by a straightforward, yet very time intensive extension of the current approach.</p><p>The results obtained in the present remodeling study prove the suitability of our approach for interdomain-angle optimization. In addition, the high prediction rate obtained specifically for the energetically highest ranked poses further demonstrates that our method is a powerful candidate for blind prediction. Therefore it should be well suited as part of any accurate atomistic modeling pipeline for TCRpMHC complexes and potentially other large molecular assemblies.</p>\",\"PeriodicalId\":498,\"journal\":{\"name\":\"BMC Structural Biology\",\"volume\":\"17 1\",\"pages\":\"\"},\"PeriodicalIF\":2.2220,\"publicationDate\":\"2017-02-02\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/s12900-016-0071-7\",\"citationCount\":\"11\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"BMC Structural Biology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://link.springer.com/article/10.1186/s12900-016-0071-7\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"Biochemistry, Genetics and Molecular Biology\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Structural Biology","FirstCategoryId":"1085","ListUrlMain":"https://link.springer.com/article/10.1186/s12900-016-0071-7","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
DynaDom: structure-based prediction of T cell receptor inter-domain and T cell receptor-peptide-MHC (class I) association angles
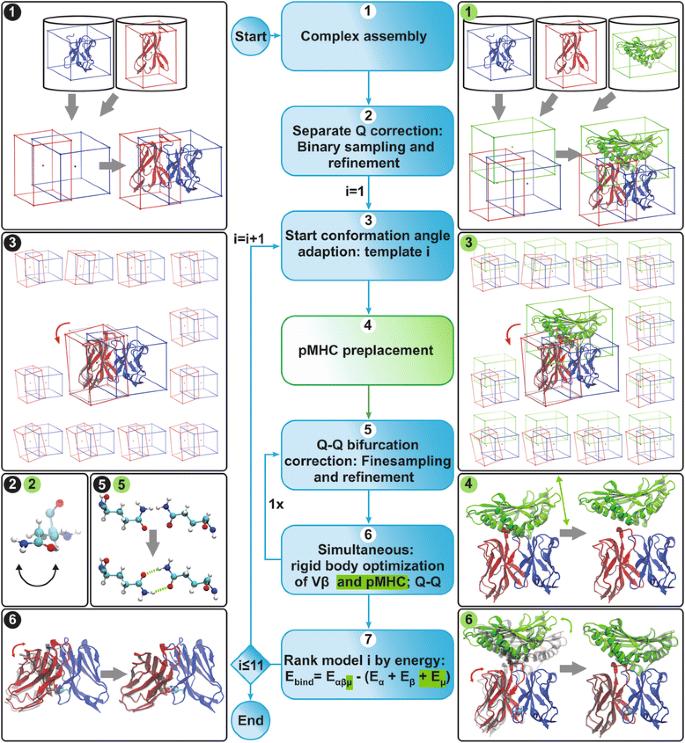
T cell receptor (TCR) molecules are involved in the adaptive immune response as they distinguish between self- and foreign-peptides, presented in major histocompatibility complex molecules (pMHC). Former studies showed that the association angles of the TCR variable domains (Vα/Vβ) can differ significantly and change upon binding to the pMHC complex. These changes can be described as a rotation of the domains around a general Center of Rotation, characterized by the interaction of two highly conserved glutamine residues.
We developed a computational method, DynaDom, for the prediction of TCR Vα/Vβ inter-domain and TCR/pMHC orientations in TCRpMHC complexes, which allows predicting the orientation of multiple protein-domains. In addition, we implemented a new approach to predict the correct orientation of the carboxamide endgroups in glutamine and asparagine residues, which can also be used as an external, independent tool.
The approach was evaluated for the remodeling of 75 and 53 experimental structures of TCR and TCRpMHC (class I) complexes, respectively. We show that the DynaDom method predicts the correct orientation of the TCR Vα/Vβ angles in 96 and 89% of the cases, for the poses with the best RMSD and best interaction energy, respectively. For the concurrent prediction of the TCR Vα/Vβ and pMHC orientations, the respective rates reached 74 and 72%. Through an exhaustive analysis, we could show that the pMHC placement can be further improved by a straightforward, yet very time intensive extension of the current approach.
The results obtained in the present remodeling study prove the suitability of our approach for interdomain-angle optimization. In addition, the high prediction rate obtained specifically for the energetically highest ranked poses further demonstrates that our method is a powerful candidate for blind prediction. Therefore it should be well suited as part of any accurate atomistic modeling pipeline for TCRpMHC complexes and potentially other large molecular assemblies.
期刊介绍:
BMC Structural Biology is an open access, peer-reviewed journal that considers articles on investigations into the structure of biological macromolecules, including solving structures, structural and functional analyses, and computational modeling.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


