{"title":"泰国甲型流感病毒的基因组特征和突变分析。","authors":"Somruthai Rattanaburi, Vorthon Sawaswong, Pattaraporn Nimsamer, Oraphan Mayuramart, Pavaret Sivapornnukul, Ariya Khamwut, Prangwalai Chanchaem, Kritsada Kongnomnan, Nungruthai Suntronwong, Yong Poovorawan, Sunchai Payungporn","doi":"10.5808/gi.21077","DOIUrl":null,"url":null,"abstract":"<p><p>The influenza A viruses have high mutation rates and cause a serious health problem worldwide. Therefore, this study focused on genome characterization of the viruses isolated from Thai patients based on the next-generation sequencing technology. The nasal swabs were collected from patients with influenza-like illness in Thailand during 2017-2018. Then, the influenza A viruses were detected by reverse transcription-quantitative polymerase chain reaction and isolated by MDCK cells. The viral genomes were amplified and sequenced by Illumina MiSeq platform. Whole genome sequences were used for characterization, phylogenetic construction, mutation analysis and nucleotide diversity of the viruses. The result revealed that 90 samples were positive for the viruses including 44 of A/ H1N1 and 46 of A/H3N2. Among these, 43 samples were successfully isolated and then the viral genomes of 25 samples were completely amplified. Finally, 17 whole genomes of the viruses (A/H1N1, n=12 and A/H3N2, n=5) were successfully sequenced with an average of 232,578 mapped reads and 1,720 genome coverage per sample. Phylogenetic analysis demonstrated that the A/H1N1 viruses were distinguishable from the recommended vaccine strains. However, the A/H3N2 viruses from this study were closely related to the recommended vaccine strains. The nonsynonymous mutations were found in all genes of both viruses, especially in hemagglutinin (HA) and neuraminidase (NA) genes. The nucleotide diversity analysis revealed negative selection in the PB1, PA, HA, and NA genes of the A/H1N1 viruses. High-throughput data in this study allow for genetic characterization of circulating influenza viruses which would be crucial for preparation against pandemic and epidemic outbreaks in the future.</p>","PeriodicalId":36591,"journal":{"name":"Genomics and Informatics","volume":"20 2","pages":"e21"},"PeriodicalIF":0.0000,"publicationDate":"2022-06-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9299564/pdf/","citationCount":"1","resultStr":"{\"title\":\"Genome characterization and mutation analysis of human influenza A virus in Thailand.\",\"authors\":\"Somruthai Rattanaburi, Vorthon Sawaswong, Pattaraporn Nimsamer, Oraphan Mayuramart, Pavaret Sivapornnukul, Ariya Khamwut, Prangwalai Chanchaem, Kritsada Kongnomnan, Nungruthai Suntronwong, Yong Poovorawan, Sunchai Payungporn\",\"doi\":\"10.5808/gi.21077\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>The influenza A viruses have high mutation rates and cause a serious health problem worldwide. Therefore, this study focused on genome characterization of the viruses isolated from Thai patients based on the next-generation sequencing technology. The nasal swabs were collected from patients with influenza-like illness in Thailand during 2017-2018. Then, the influenza A viruses were detected by reverse transcription-quantitative polymerase chain reaction and isolated by MDCK cells. The viral genomes were amplified and sequenced by Illumina MiSeq platform. Whole genome sequences were used for characterization, phylogenetic construction, mutation analysis and nucleotide diversity of the viruses. The result revealed that 90 samples were positive for the viruses including 44 of A/ H1N1 and 46 of A/H3N2. Among these, 43 samples were successfully isolated and then the viral genomes of 25 samples were completely amplified. Finally, 17 whole genomes of the viruses (A/H1N1, n=12 and A/H3N2, n=5) were successfully sequenced with an average of 232,578 mapped reads and 1,720 genome coverage per sample. Phylogenetic analysis demonstrated that the A/H1N1 viruses were distinguishable from the recommended vaccine strains. However, the A/H3N2 viruses from this study were closely related to the recommended vaccine strains. The nonsynonymous mutations were found in all genes of both viruses, especially in hemagglutinin (HA) and neuraminidase (NA) genes. The nucleotide diversity analysis revealed negative selection in the PB1, PA, HA, and NA genes of the A/H1N1 viruses. High-throughput data in this study allow for genetic characterization of circulating influenza viruses which would be crucial for preparation against pandemic and epidemic outbreaks in the future.</p>\",\"PeriodicalId\":36591,\"journal\":{\"name\":\"Genomics and Informatics\",\"volume\":\"20 2\",\"pages\":\"e21\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2022-06-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9299564/pdf/\",\"citationCount\":\"1\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Genomics and Informatics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.5808/gi.21077\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2022/6/30 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"Agricultural and Biological Sciences\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Genomics and Informatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.5808/gi.21077","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/6/30 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"Agricultural and Biological Sciences","Score":null,"Total":0}
Genome characterization and mutation analysis of human influenza A virus in Thailand.
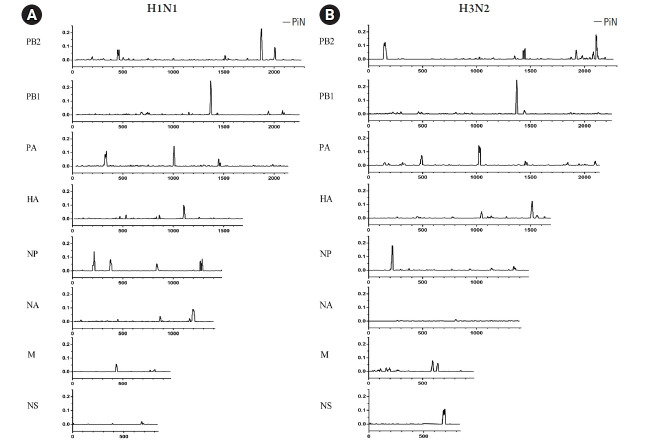
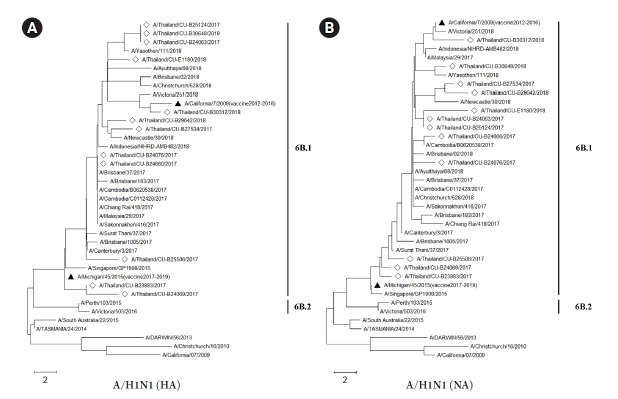
The influenza A viruses have high mutation rates and cause a serious health problem worldwide. Therefore, this study focused on genome characterization of the viruses isolated from Thai patients based on the next-generation sequencing technology. The nasal swabs were collected from patients with influenza-like illness in Thailand during 2017-2018. Then, the influenza A viruses were detected by reverse transcription-quantitative polymerase chain reaction and isolated by MDCK cells. The viral genomes were amplified and sequenced by Illumina MiSeq platform. Whole genome sequences were used for characterization, phylogenetic construction, mutation analysis and nucleotide diversity of the viruses. The result revealed that 90 samples were positive for the viruses including 44 of A/ H1N1 and 46 of A/H3N2. Among these, 43 samples were successfully isolated and then the viral genomes of 25 samples were completely amplified. Finally, 17 whole genomes of the viruses (A/H1N1, n=12 and A/H3N2, n=5) were successfully sequenced with an average of 232,578 mapped reads and 1,720 genome coverage per sample. Phylogenetic analysis demonstrated that the A/H1N1 viruses were distinguishable from the recommended vaccine strains. However, the A/H3N2 viruses from this study were closely related to the recommended vaccine strains. The nonsynonymous mutations were found in all genes of both viruses, especially in hemagglutinin (HA) and neuraminidase (NA) genes. The nucleotide diversity analysis revealed negative selection in the PB1, PA, HA, and NA genes of the A/H1N1 viruses. High-throughput data in this study allow for genetic characterization of circulating influenza viruses which would be crucial for preparation against pandemic and epidemic outbreaks in the future.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


