{"title":"先天性肌病与MEGF10基因的新突变、肌原纤维改变和进展过程相关。","authors":"Carolina Croci, Monica Traverso, Serena Baratto, Michele Iacomino, Marina Pedemonte, Francesco Caroli, Marcello Scala, Claudio Bruno, Chiara Fiorillo","doi":"10.36185/2532-1900-076","DOIUrl":null,"url":null,"abstract":"<p><p>Early-onset myopathy, areflexia, respiratory distress, and dysphagia (EMARDD) is caused by homozygous or compound heterozygous mutation in the <i>MEGF10</i> gene (OMIM #614399). Phenotypic spectrum of EMARDD is variable, ranging from severe infantile forms in which patients are ventilator-dependent and die in childhood, to milder chronic disorders with a more favorable course (mild variant, mvEMARDD). Here we describe a 22 years old boy, offspring of consanguineous parents, presenting a congenital myopathic phenotype since infancy with elbow contractures and scoliosis. The patient developed a slowly progressive muscle weakness with impaired walking, rhinolalia, dysphagia, and respiratory involvement, which required noninvasive ventilation therapy since the age of 16 years. First muscle biopsy revealed unspecific muscle damage, with fiber size variation, internal nuclei and fibrosis. Myofibrillar alterations were noted at a second muscle biopsy including whorled fibres, cytoplasmic inclusion and minicores. Exome sequencing identified a homozygous mutation in <i>MEGF10</i> gene, c.2096G > C (p.Cys699Ser), inherited by both parents. This variant, not reported in public databases of mutations, is expected to alter the structure of the protein and is therefore predicted to be probably damaging according to ACMG classification. In conclusion, we found a new likely pathogenic mutation in <i>MEGF10</i>, which is responsible for a progressive form of mvEMARDD with myofibrillar alterations at muscle biopsy. Interestingly, the presence of <i>MEGF10</i> mutations has not been reported in Italian population. Early diagnosis of MEGF10 myopathy is essential in light of recent results from in vivo testing demonstrating a potential therapeutic effect of SSRIs compounds.</p>","PeriodicalId":35953,"journal":{"name":"Acta Myologica","volume":"41 3","pages":"111-116"},"PeriodicalIF":0.0000,"publicationDate":"2022-09-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/34/97/am-2022-03-111.PMC9628799.pdf","citationCount":"0","resultStr":"{\"title\":\"Congenital myopathy associated with a novel mutation in <i>MEGF10</i> gene, myofibrillar alteration and progressive course.\",\"authors\":\"Carolina Croci, Monica Traverso, Serena Baratto, Michele Iacomino, Marina Pedemonte, Francesco Caroli, Marcello Scala, Claudio Bruno, Chiara Fiorillo\",\"doi\":\"10.36185/2532-1900-076\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Early-onset myopathy, areflexia, respiratory distress, and dysphagia (EMARDD) is caused by homozygous or compound heterozygous mutation in the <i>MEGF10</i> gene (OMIM #614399). Phenotypic spectrum of EMARDD is variable, ranging from severe infantile forms in which patients are ventilator-dependent and die in childhood, to milder chronic disorders with a more favorable course (mild variant, mvEMARDD). Here we describe a 22 years old boy, offspring of consanguineous parents, presenting a congenital myopathic phenotype since infancy with elbow contractures and scoliosis. The patient developed a slowly progressive muscle weakness with impaired walking, rhinolalia, dysphagia, and respiratory involvement, which required noninvasive ventilation therapy since the age of 16 years. First muscle biopsy revealed unspecific muscle damage, with fiber size variation, internal nuclei and fibrosis. Myofibrillar alterations were noted at a second muscle biopsy including whorled fibres, cytoplasmic inclusion and minicores. Exome sequencing identified a homozygous mutation in <i>MEGF10</i> gene, c.2096G > C (p.Cys699Ser), inherited by both parents. This variant, not reported in public databases of mutations, is expected to alter the structure of the protein and is therefore predicted to be probably damaging according to ACMG classification. In conclusion, we found a new likely pathogenic mutation in <i>MEGF10</i>, which is responsible for a progressive form of mvEMARDD with myofibrillar alterations at muscle biopsy. Interestingly, the presence of <i>MEGF10</i> mutations has not been reported in Italian population. Early diagnosis of MEGF10 myopathy is essential in light of recent results from in vivo testing demonstrating a potential therapeutic effect of SSRIs compounds.</p>\",\"PeriodicalId\":35953,\"journal\":{\"name\":\"Acta Myologica\",\"volume\":\"41 3\",\"pages\":\"111-116\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2022-09-30\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/34/97/am-2022-03-111.PMC9628799.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Acta Myologica\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.36185/2532-1900-076\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2022/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q3\",\"JCRName\":\"Medicine\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Acta Myologica","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.36185/2532-1900-076","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/1/1 0:00:00","PubModel":"eCollection","JCR":"Q3","JCRName":"Medicine","Score":null,"Total":0}
引用次数: 0
摘要
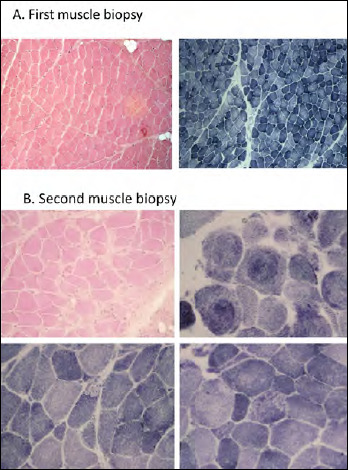
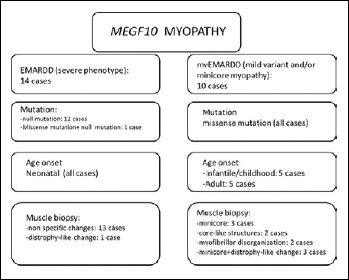
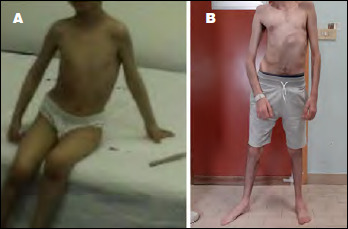
早发性肌病、反射性松弛、呼吸窘迫和吞咽困难(EMARDD)是由MEGF10基因(OMIM #614399)的纯合或复合杂合突变引起的。EMARDD的表型谱是可变的,从严重的婴儿型(患者依赖呼吸机并在儿童期死亡)到较轻的慢性疾病(轻度变异,mvEMARDD),病程较好。在这里,我们描述了一个22岁的男孩,近亲父母的后代,表现出先天性肌病表型自婴儿期肘关节挛缩和脊柱侧凸。患者出现缓慢进行性肌肉无力,伴有行走障碍、鼻鸣、吞咽困难和呼吸受累,自16岁起需要无创通气治疗。第一次肌肉活检显示非特异性肌肉损伤,纤维大小变化,内核和纤维化。在第二次肌肉活检中发现肌纤维改变,包括螺旋纤维,胞质包涵体和微孔。外显子组测序发现MEGF10基因C . 2096g > C (p.Cys699Ser)纯合突变,由父母双方遗传。该变异未在公共突变数据库中报道,预计会改变蛋白质的结构,因此根据ACMG分类预测可能具有破坏性。总之,我们在MEGF10中发现了一种新的可能的致病突变,该突变负责肌肉活检中伴有肌纤维改变的进行性mvEMARDD。有趣的是,在意大利人群中没有MEGF10突变的报道。鉴于最近体内试验结果显示SSRIs化合物具有潜在的治疗作用,MEGF10肌病的早期诊断至关重要。
Congenital myopathy associated with a novel mutation in MEGF10 gene, myofibrillar alteration and progressive course.
Early-onset myopathy, areflexia, respiratory distress, and dysphagia (EMARDD) is caused by homozygous or compound heterozygous mutation in the MEGF10 gene (OMIM #614399). Phenotypic spectrum of EMARDD is variable, ranging from severe infantile forms in which patients are ventilator-dependent and die in childhood, to milder chronic disorders with a more favorable course (mild variant, mvEMARDD). Here we describe a 22 years old boy, offspring of consanguineous parents, presenting a congenital myopathic phenotype since infancy with elbow contractures and scoliosis. The patient developed a slowly progressive muscle weakness with impaired walking, rhinolalia, dysphagia, and respiratory involvement, which required noninvasive ventilation therapy since the age of 16 years. First muscle biopsy revealed unspecific muscle damage, with fiber size variation, internal nuclei and fibrosis. Myofibrillar alterations were noted at a second muscle biopsy including whorled fibres, cytoplasmic inclusion and minicores. Exome sequencing identified a homozygous mutation in MEGF10 gene, c.2096G > C (p.Cys699Ser), inherited by both parents. This variant, not reported in public databases of mutations, is expected to alter the structure of the protein and is therefore predicted to be probably damaging according to ACMG classification. In conclusion, we found a new likely pathogenic mutation in MEGF10, which is responsible for a progressive form of mvEMARDD with myofibrillar alterations at muscle biopsy. Interestingly, the presence of MEGF10 mutations has not been reported in Italian population. Early diagnosis of MEGF10 myopathy is essential in light of recent results from in vivo testing demonstrating a potential therapeutic effect of SSRIs compounds.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


