Ludovica Pasca, Alice Gardani, Matteo Paoletti, Daniele Velardo, Angela Berardinelli
{"title":"一名患有杜氏肌营养不良症的男孩晚期用阿特洛仑治疗后反应良好:以前的轻度病程会影响结果吗?","authors":"Ludovica Pasca, Alice Gardani, Matteo Paoletti, Daniele Velardo, Angela Berardinelli","doi":"10.36185/2532-1900-078","DOIUrl":null,"url":null,"abstract":"<p><p>Duchenne muscular dystrophy (DMD) is a severe, progressive X-linked recessive disorder, caused by the absence of the dystrophin protein. A resolutive therapy for DMD is not yet available. The first approved drug for DMD patients with nonsense mutations is ataluren, approved for the treatment of children aged ≥ 2 yrs, that seems effective in slowing the disease progression. An earlier introduction of ataluren seems to give better results. We report the case of a 14-year-old DMD patient with a nonsense mutation in exon 70, still ambulant, who started taking ataluren at 12 years and remained stable for the following two years. The patient was on steroid since the age of 6, with beneficial effects. At two-years follow-up, an optimal disease evolution was observed, associated with a constant decrease of creatine kinase blood levels. Despite the late start of the treatment, ataluren seems to have significantly contributed to the stabilization of the functional status in this patient though it cannot be excluded that the result may have been influenced by the previous favorable course of the disease. However, further studies should be planned in patients with similar age treated with ataluren to better evaluate the treatment's results compared to the natural course of the disease.</p>","PeriodicalId":35953,"journal":{"name":"Acta Myologica","volume":"41 3","pages":"121-125"},"PeriodicalIF":0.0000,"publicationDate":"2022-09-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/93/fa/am-2022-03-121.PMC9628801.pdf","citationCount":"1","resultStr":"{\"title\":\"Good response to the late treatment with ataluren in a boy with Duchenne muscular dystrophy: could the previous mild course of the disease have affected the outcome?\",\"authors\":\"Ludovica Pasca, Alice Gardani, Matteo Paoletti, Daniele Velardo, Angela Berardinelli\",\"doi\":\"10.36185/2532-1900-078\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Duchenne muscular dystrophy (DMD) is a severe, progressive X-linked recessive disorder, caused by the absence of the dystrophin protein. A resolutive therapy for DMD is not yet available. The first approved drug for DMD patients with nonsense mutations is ataluren, approved for the treatment of children aged ≥ 2 yrs, that seems effective in slowing the disease progression. An earlier introduction of ataluren seems to give better results. We report the case of a 14-year-old DMD patient with a nonsense mutation in exon 70, still ambulant, who started taking ataluren at 12 years and remained stable for the following two years. The patient was on steroid since the age of 6, with beneficial effects. At two-years follow-up, an optimal disease evolution was observed, associated with a constant decrease of creatine kinase blood levels. Despite the late start of the treatment, ataluren seems to have significantly contributed to the stabilization of the functional status in this patient though it cannot be excluded that the result may have been influenced by the previous favorable course of the disease. However, further studies should be planned in patients with similar age treated with ataluren to better evaluate the treatment's results compared to the natural course of the disease.</p>\",\"PeriodicalId\":35953,\"journal\":{\"name\":\"Acta Myologica\",\"volume\":\"41 3\",\"pages\":\"121-125\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2022-09-30\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/93/fa/am-2022-03-121.PMC9628801.pdf\",\"citationCount\":\"1\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Acta Myologica\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.36185/2532-1900-078\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2022/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q3\",\"JCRName\":\"Medicine\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Acta Myologica","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.36185/2532-1900-078","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/1/1 0:00:00","PubModel":"eCollection","JCR":"Q3","JCRName":"Medicine","Score":null,"Total":0}
Good response to the late treatment with ataluren in a boy with Duchenne muscular dystrophy: could the previous mild course of the disease have affected the outcome?
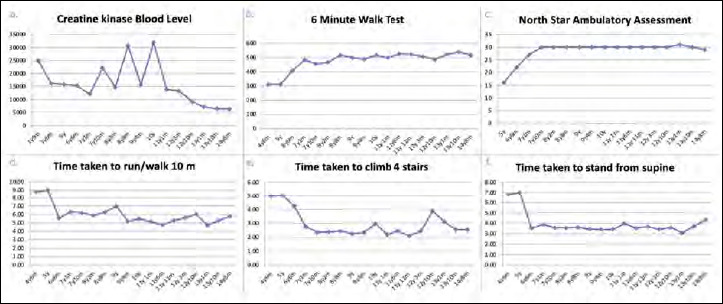
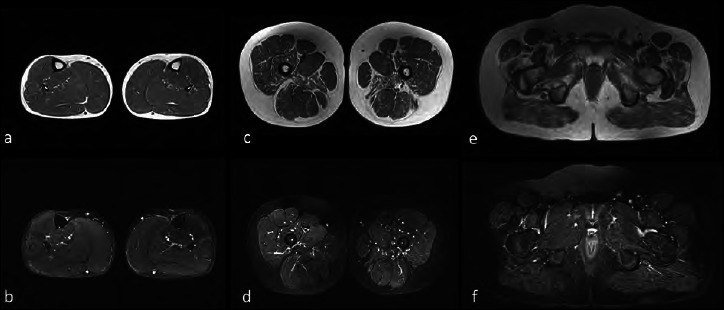
Duchenne muscular dystrophy (DMD) is a severe, progressive X-linked recessive disorder, caused by the absence of the dystrophin protein. A resolutive therapy for DMD is not yet available. The first approved drug for DMD patients with nonsense mutations is ataluren, approved for the treatment of children aged ≥ 2 yrs, that seems effective in slowing the disease progression. An earlier introduction of ataluren seems to give better results. We report the case of a 14-year-old DMD patient with a nonsense mutation in exon 70, still ambulant, who started taking ataluren at 12 years and remained stable for the following two years. The patient was on steroid since the age of 6, with beneficial effects. At two-years follow-up, an optimal disease evolution was observed, associated with a constant decrease of creatine kinase blood levels. Despite the late start of the treatment, ataluren seems to have significantly contributed to the stabilization of the functional status in this patient though it cannot be excluded that the result may have been influenced by the previous favorable course of the disease. However, further studies should be planned in patients with similar age treated with ataluren to better evaluate the treatment's results compared to the natural course of the disease.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


