GREPore-seq:通过长程 PCR 和纳米孔测序检测基因编辑后变化的强大工作流程。
IF 7.9
2区 生物学
Q1 GENETICS & HEREDITY
引用次数: 0
摘要
为了发挥基因编辑技术在临床治疗中的巨大潜力,我们需要全面评估靶向效率和意外编辑后果。然而,目前还缺乏一种流水线式、大规模且经济的工作流程来检测基因组编辑结果,尤其是大片段的插入或缺失。在这里,我们介绍了一种通过对条形码长程 PCR 产物进行集合纳米孔测序,高效准确地检测 CRISPR/Cas9 编辑后多种基因变化的方法。由于认识到牛津纳米孔测序的高错误率,我们开发了一种新型管道,通过对纳米孔扩增子测序的读数进行抓取(GREPore-seq)来捕获条形码序列。GREPore-seq 可以评估非同源末端连接(NHEJ)介导的双链寡脱氧核苷酸(dsODN)插入,其准确性与 Illumina 下一代测序(NGS)相当。GREPore-seq 还揭示了同源定向修复(HDR)介导的大基因敲入的全谱,与荧光激活细胞分选(FACS)分析结果密切相关。值得注意的是,我们在 CRISPR 切割位点发现了低水平的片段和全长质粒骨架插入。因此,我们建立了一套实用的工作流程来评估各种基因变化,包括量化 CRISPR 编辑后的短 dsODNs 插入、长片段敲入、质粒插入和大片段缺失。GREPore-seq可在GitHub(https://github.com/lisiang/GREPore-seq)和美国国家基因组学数据中心(NGDC)生物代码(https://ngdc.cncb.ac.cn/biocode/tools/BT007293)上免费获取。本文章由计算机程序翻译,如有差异,请以英文原文为准。
GREPore-seq: A Robust Workflow to Detect Changes After Gene Editing Through Long-range PCR and Nanopore Sequencing
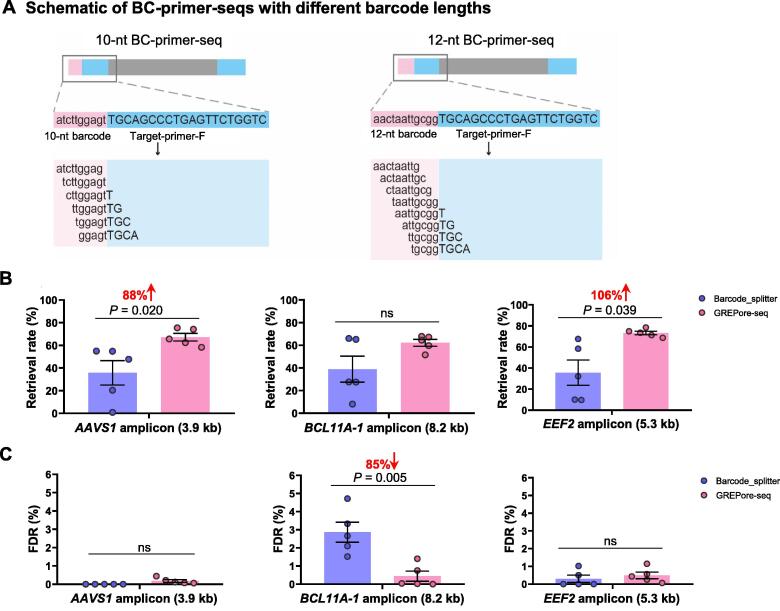
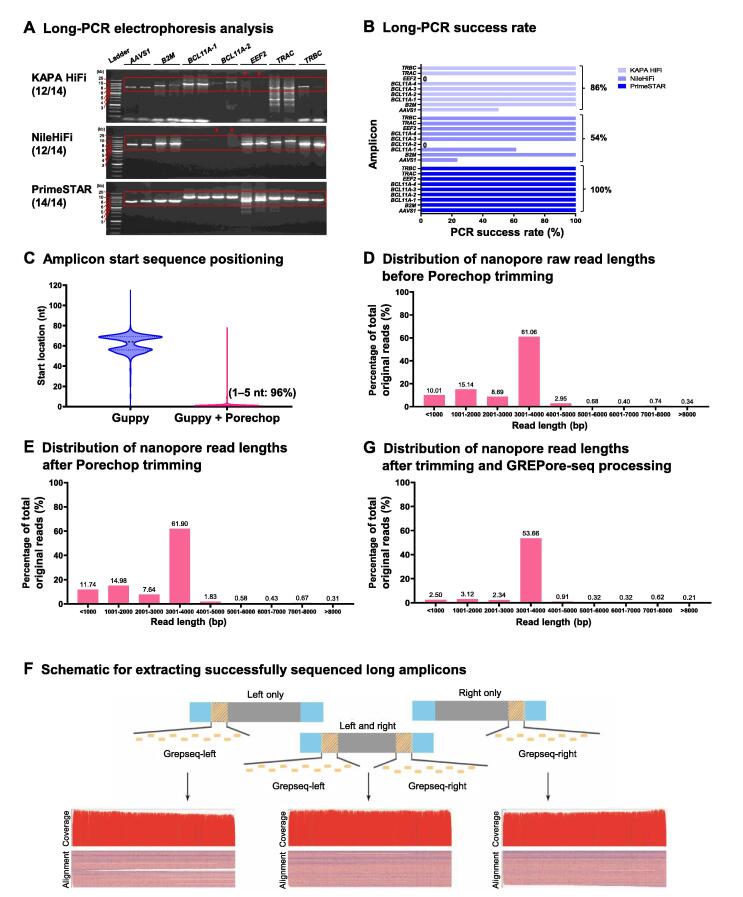
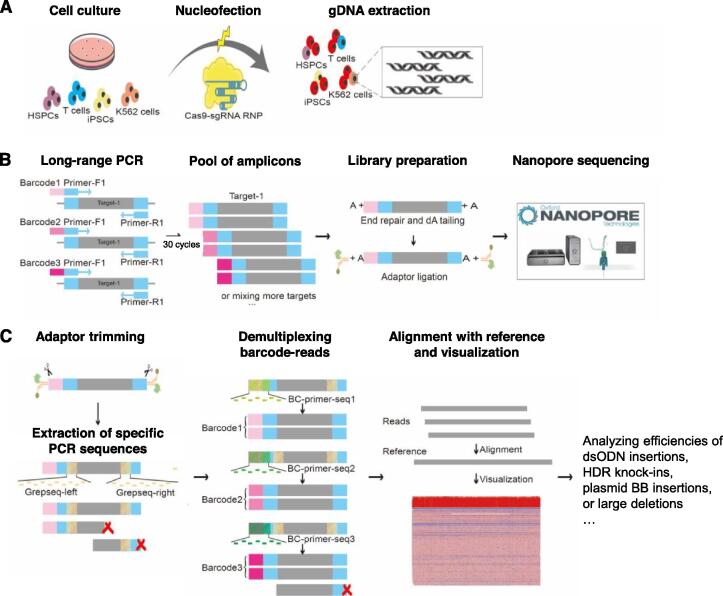
To achieve the enormous potential of gene-editing technology in clinical therapies, one needs to evaluate both the on-target efficiency and unintended editing consequences comprehensively. However, there is a lack of a pipelined, large-scale, and economical workflow for detecting genome editing outcomes, in particular insertion or deletion of a large fragment. Here, we describe an approach for efficient and accurate detection of multiple genetic changes after CRISPR/Cas9 editing by pooled nanopore sequencing of barcoded long-range PCR products. Recognizing the high error rates of Oxford nanopore sequencing, we developed a novel pipeline to capture the barcoded sequences by grepping reads of nanopore amplicon sequencing (GREPore-seq). GREPore-seq can assess nonhomologous end-joining (NHEJ)-mediated double-stranded oligodeoxynucleotide (dsODN) insertions with comparable accuracy to Illumina next-generation sequencing (NGS). GREPore-seq also reveals a full spectrum of homology-directed repair (HDR)-mediated large gene knock-in, correlating well with the fluorescence-activated cell sorting (FACS) analysis results. Of note, we discovered low-level fragmented and full-length plasmid backbone insertion at the CRISPR cutting site. Therefore, we have established a practical workflow to evaluate various genetic changes, including quantifying insertions of short dsODNs, knock-ins of long pieces, plasmid insertions, and large fragment deletions after CRISPR/Cas9-mediated editing. GREPore-seq is freely available at GitHub (https://github.com/lisiang/GREPore-seq) and the National Genomics Data Center (NGDC) BioCode (https://ngdc.cncb.ac.cn/biocode/tools/BT007293).
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Genomics, Proteomics & Bioinformatics
Biochemistry, Genetics and Molecular Biology-Biochemistry
CiteScore
14.30
自引率
4.20%
发文量
844
审稿时长
61 days
期刊介绍:
Genomics, Proteomics and Bioinformatics (GPB) is the official journal of the Beijing Institute of Genomics, Chinese Academy of Sciences / China National Center for Bioinformation and Genetics Society of China. It aims to disseminate new developments in the field of omics and bioinformatics, publish high-quality discoveries quickly, and promote open access and online publication. GPB welcomes submissions in all areas of life science, biology, and biomedicine, with a focus on large data acquisition, analysis, and curation. Manuscripts covering omics and related bioinformatics topics are particularly encouraged. GPB is indexed/abstracted by PubMed/MEDLINE, PubMed Central, Scopus, BIOSIS Previews, Chemical Abstracts, CSCD, among others.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


