{"title":"特发性肺纤维化、结缔组织病相关间质性肺病、间质性肺炎伴自身免疫性特征的临床、功能和预后评价:一项单中心前瞻性研究","authors":"Miraç Öz, Serhat Erol, Orhan Küçükşahin, İrem Kar, Kayhan Çetin Atasoy, Özlem Özdemir Kumbasar","doi":"10.5152/TurkThoracJ.2022.22017","DOIUrl":null,"url":null,"abstract":"<p><strong>Objective: </strong>Our study aimed to evaluate clinical, functional, and prognostic features and to determine the prognosis of idiopathic pulmonary fibrosis, connective tissue disease-associated interstitial lung diseases, and interstitial pneumonia with autoimmune features.</p><p><strong>Material and methods: </strong>Sixty-nine cases with interstitial lung diseases were recruited in this study prospectively. Demographic features, symptoms, radiological findings, functional measurements, and immunological markers were recorded twice (at the time of initial admission and in the 12th month). Twenty-four of 69 cases were idiopathic pulmonary fibrosis, 32 were connective tissue diseaseassociated interstitial lung diseases, and 13 were interstitial pneumonia with autoimmune features .</p><p><strong>Results: </strong>Most of the patients with idiopathic pulmonary fibrosis were male, while there were more female patients in connective tissue disease-associated interstitial lung diseases and interstitial pneumonia with autoimmune features groups. Female patients (65.0%) predominated in connective tissue disease-associated interstitial lung diseases group (P <.001). There was no significant difference in the mean ages of the disease groups, yet connective tissue disease-associated interstitial lung diseases patients were generally younger (min- max: 34-82 years). In the idiopathic pulmonary fibrosis group, only low titers of antinuclear antibody positivity were found. Antinuclear antibody positivity in the connective tissue disease-associated interstitial lung diseases group and interstitial pneumonia with autoimmune features group was high (P = .001). The long-term survival of idiopathic pulmonary fibrosis, connective tissue disease-associated interstitial lung diseases, and interstitial pneumonia with autoimmune features patients were 37%, 40 months (median) (95% CI, 5.193- 74.807), 48.6%, 80 months (median) (95% CI, 57.032-102.968), 30.8%, 46 months (median) (95% CI, 26.624-65.376), respectively.</p><p><strong>Conclusion: </strong>Although a consensus report describing interstitial lung diseases with autoimmune features has been published, diagnostic criteria for this group are still vague. Since the interstitial pneumonia with autoimmune features group had the worst results in terms of functional loss and survival rates, the follow-up parameters and follow-up algorithm should be established for this group. Clinical and immunological evaluation of the interstitial pneumonia with autoimmune features group should include detailed parameters because of follow-up and to estimate survival.</p>","PeriodicalId":37452,"journal":{"name":"Turkish Thoracic Journal","volume":" ","pages":"395-402"},"PeriodicalIF":0.8000,"publicationDate":"2022-11-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/2f/32/ttj-23-6-395.PMC9682965.pdf","citationCount":"0","resultStr":"{\"title\":\"Clinical, Functional, and Prognostic Evaluation of Idiopathic Pulmonary Fibrosis, Connective Tissue Disease-Associated Interstitial Lung Disease, Interstitial Pneumonia with Autoimmune Features: A Single-Center Prospective Study.\",\"authors\":\"Miraç Öz, Serhat Erol, Orhan Küçükşahin, İrem Kar, Kayhan Çetin Atasoy, Özlem Özdemir Kumbasar\",\"doi\":\"10.5152/TurkThoracJ.2022.22017\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Objective: </strong>Our study aimed to evaluate clinical, functional, and prognostic features and to determine the prognosis of idiopathic pulmonary fibrosis, connective tissue disease-associated interstitial lung diseases, and interstitial pneumonia with autoimmune features.</p><p><strong>Material and methods: </strong>Sixty-nine cases with interstitial lung diseases were recruited in this study prospectively. Demographic features, symptoms, radiological findings, functional measurements, and immunological markers were recorded twice (at the time of initial admission and in the 12th month). Twenty-four of 69 cases were idiopathic pulmonary fibrosis, 32 were connective tissue diseaseassociated interstitial lung diseases, and 13 were interstitial pneumonia with autoimmune features .</p><p><strong>Results: </strong>Most of the patients with idiopathic pulmonary fibrosis were male, while there were more female patients in connective tissue disease-associated interstitial lung diseases and interstitial pneumonia with autoimmune features groups. Female patients (65.0%) predominated in connective tissue disease-associated interstitial lung diseases group (P <.001). There was no significant difference in the mean ages of the disease groups, yet connective tissue disease-associated interstitial lung diseases patients were generally younger (min- max: 34-82 years). In the idiopathic pulmonary fibrosis group, only low titers of antinuclear antibody positivity were found. Antinuclear antibody positivity in the connective tissue disease-associated interstitial lung diseases group and interstitial pneumonia with autoimmune features group was high (P = .001). The long-term survival of idiopathic pulmonary fibrosis, connective tissue disease-associated interstitial lung diseases, and interstitial pneumonia with autoimmune features patients were 37%, 40 months (median) (95% CI, 5.193- 74.807), 48.6%, 80 months (median) (95% CI, 57.032-102.968), 30.8%, 46 months (median) (95% CI, 26.624-65.376), respectively.</p><p><strong>Conclusion: </strong>Although a consensus report describing interstitial lung diseases with autoimmune features has been published, diagnostic criteria for this group are still vague. Since the interstitial pneumonia with autoimmune features group had the worst results in terms of functional loss and survival rates, the follow-up parameters and follow-up algorithm should be established for this group. Clinical and immunological evaluation of the interstitial pneumonia with autoimmune features group should include detailed parameters because of follow-up and to estimate survival.</p>\",\"PeriodicalId\":37452,\"journal\":{\"name\":\"Turkish Thoracic Journal\",\"volume\":\" \",\"pages\":\"395-402\"},\"PeriodicalIF\":0.8000,\"publicationDate\":\"2022-11-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/2f/32/ttj-23-6-395.PMC9682965.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Turkish Thoracic Journal\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.5152/TurkThoracJ.2022.22017\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"RESPIRATORY SYSTEM\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Turkish Thoracic Journal","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.5152/TurkThoracJ.2022.22017","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"RESPIRATORY SYSTEM","Score":null,"Total":0}
Clinical, Functional, and Prognostic Evaluation of Idiopathic Pulmonary Fibrosis, Connective Tissue Disease-Associated Interstitial Lung Disease, Interstitial Pneumonia with Autoimmune Features: A Single-Center Prospective Study.
Objective: Our study aimed to evaluate clinical, functional, and prognostic features and to determine the prognosis of idiopathic pulmonary fibrosis, connective tissue disease-associated interstitial lung diseases, and interstitial pneumonia with autoimmune features.
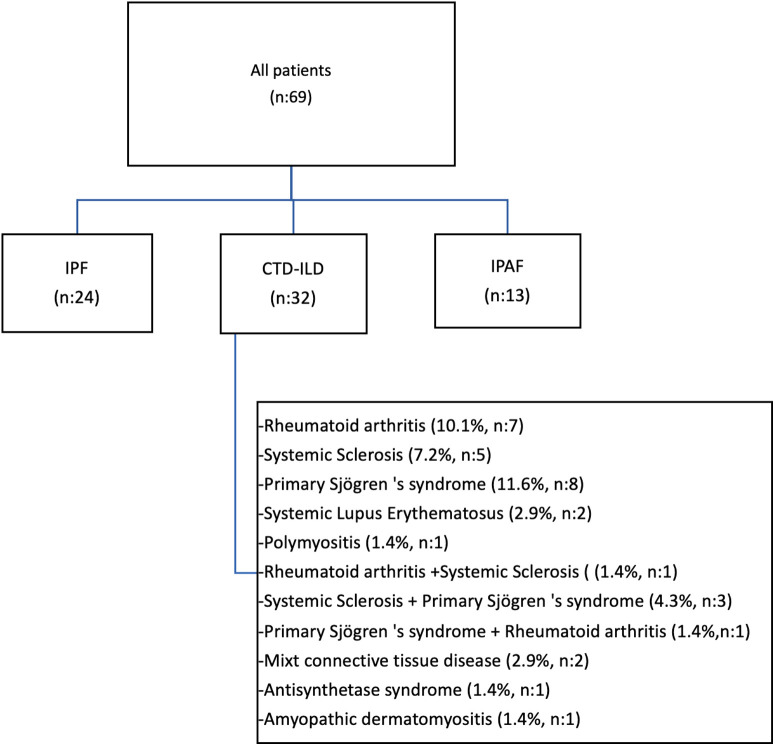
Material and methods: Sixty-nine cases with interstitial lung diseases were recruited in this study prospectively. Demographic features, symptoms, radiological findings, functional measurements, and immunological markers were recorded twice (at the time of initial admission and in the 12th month). Twenty-four of 69 cases were idiopathic pulmonary fibrosis, 32 were connective tissue diseaseassociated interstitial lung diseases, and 13 were interstitial pneumonia with autoimmune features .
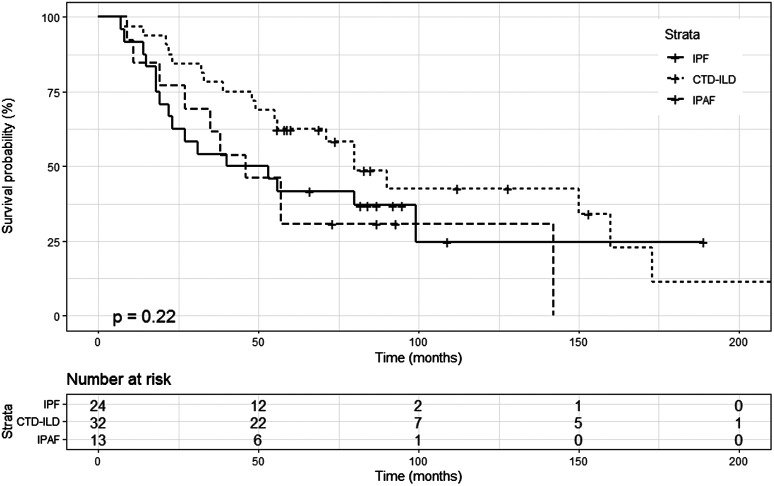
Results: Most of the patients with idiopathic pulmonary fibrosis were male, while there were more female patients in connective tissue disease-associated interstitial lung diseases and interstitial pneumonia with autoimmune features groups. Female patients (65.0%) predominated in connective tissue disease-associated interstitial lung diseases group (P <.001). There was no significant difference in the mean ages of the disease groups, yet connective tissue disease-associated interstitial lung diseases patients were generally younger (min- max: 34-82 years). In the idiopathic pulmonary fibrosis group, only low titers of antinuclear antibody positivity were found. Antinuclear antibody positivity in the connective tissue disease-associated interstitial lung diseases group and interstitial pneumonia with autoimmune features group was high (P = .001). The long-term survival of idiopathic pulmonary fibrosis, connective tissue disease-associated interstitial lung diseases, and interstitial pneumonia with autoimmune features patients were 37%, 40 months (median) (95% CI, 5.193- 74.807), 48.6%, 80 months (median) (95% CI, 57.032-102.968), 30.8%, 46 months (median) (95% CI, 26.624-65.376), respectively.
Conclusion: Although a consensus report describing interstitial lung diseases with autoimmune features has been published, diagnostic criteria for this group are still vague. Since the interstitial pneumonia with autoimmune features group had the worst results in terms of functional loss and survival rates, the follow-up parameters and follow-up algorithm should be established for this group. Clinical and immunological evaluation of the interstitial pneumonia with autoimmune features group should include detailed parameters because of follow-up and to estimate survival.
期刊介绍:
Turkish Thoracic Journal (Turk Thorac J) is the double-blind, peer-reviewed, open access, international publication organ of Turkish Thoracic Society. The journal is a quarterly publication, published on January, April, July, and October and its publication language is English. Turkish Thoracic Journal started its publication life following the merger of two journals which were published under the titles “Turkish Respiratory Journal” and “Toraks Journal” until 2007. Archives of both journals were passed on to the Turkish Thoracic Journal. The aim of the journal is to convey scientific developments and to create a dynamic discussion platform about pulmonary diseases. With this intent, the journal accepts articles from all related scientific areas that address adult and pediatric pulmonary diseases, as well as thoracic imaging, environmental and occupational disorders, intensive care, sleep disorders and thoracic surgery. Clinical and research articles, reviews, statements of agreement or disagreement on controversial issues, national and international consensus reports, abstracts and comments of important international articles, interesting case reports, writings related to clinical and practical applications, letters to the editor, and editorials are accepted.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


