Ari Horton, Kai Mun Hong, Dinusha Pandithan, Meredith Allen, Caroline Killick, Stacy Goergen, Amanda Springer, Dean Phelan, Melanie Marty, Rebecca Halligan, Joy Lee, James Pitt, Belinda Chong, John Christodoulou, Sebastian Lunke, Zornitza Stark, Michael Fahey
{"title":"乙基丙二酸脑病伪装成脑膜炎球菌病。","authors":"Ari Horton, Kai Mun Hong, Dinusha Pandithan, Meredith Allen, Caroline Killick, Stacy Goergen, Amanda Springer, Dean Phelan, Melanie Marty, Rebecca Halligan, Joy Lee, James Pitt, Belinda Chong, John Christodoulou, Sebastian Lunke, Zornitza Stark, Michael Fahey","doi":"10.1101/mcs.a006193","DOIUrl":null,"url":null,"abstract":"<p><p>Ethylmalonic encephalopathy (MIM #602473) is a rare autosomal recessive metabolic condition caused by biallelic variants in <i>ETHE1</i> (MIM #608451), characterized by global developmental delay, infantile hypotonia, seizures, and microvascular damage. The microvascular changes result in a pattern of relapsing spontaneous diffuse petechiae and purpura, positional acrocyanosis, and pedal edema, hemorrhagic suffusions of mucous membranes, and chronic diarrhea. Here, we describe an instructive case in which ethylmalonic encephalopathy masqueraded as meningococcal septicemia and shock. Ultrarapid whole-genome testing (time to result 60 h) and prompt biochemical analysis facilitated accurate diagnosis and counseling with rapid implementation of precision treatment for the metabolic crisis related to this condition. This case provides a timely reminder to consider rare genetic diagnoses when atypical features of more common conditions are present, with an early referral to ensure prompt biochemical and genomic diagnosis.</p>","PeriodicalId":10360,"journal":{"name":"Cold Spring Harbor Molecular Case Studies","volume":" ","pages":""},"PeriodicalIF":1.8000,"publicationDate":"2022-03-24","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/9d/aa/MCS006193Hor.PMC8958906.pdf","citationCount":"3","resultStr":"{\"title\":\"Ethylmalonic encephalopathy masquerading as meningococcemia.\",\"authors\":\"Ari Horton, Kai Mun Hong, Dinusha Pandithan, Meredith Allen, Caroline Killick, Stacy Goergen, Amanda Springer, Dean Phelan, Melanie Marty, Rebecca Halligan, Joy Lee, James Pitt, Belinda Chong, John Christodoulou, Sebastian Lunke, Zornitza Stark, Michael Fahey\",\"doi\":\"10.1101/mcs.a006193\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Ethylmalonic encephalopathy (MIM #602473) is a rare autosomal recessive metabolic condition caused by biallelic variants in <i>ETHE1</i> (MIM #608451), characterized by global developmental delay, infantile hypotonia, seizures, and microvascular damage. The microvascular changes result in a pattern of relapsing spontaneous diffuse petechiae and purpura, positional acrocyanosis, and pedal edema, hemorrhagic suffusions of mucous membranes, and chronic diarrhea. Here, we describe an instructive case in which ethylmalonic encephalopathy masqueraded as meningococcal septicemia and shock. Ultrarapid whole-genome testing (time to result 60 h) and prompt biochemical analysis facilitated accurate diagnosis and counseling with rapid implementation of precision treatment for the metabolic crisis related to this condition. This case provides a timely reminder to consider rare genetic diagnoses when atypical features of more common conditions are present, with an early referral to ensure prompt biochemical and genomic diagnosis.</p>\",\"PeriodicalId\":10360,\"journal\":{\"name\":\"Cold Spring Harbor Molecular Case Studies\",\"volume\":\" \",\"pages\":\"\"},\"PeriodicalIF\":1.8000,\"publicationDate\":\"2022-03-24\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/9d/aa/MCS006193Hor.PMC8958906.pdf\",\"citationCount\":\"3\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Cold Spring Harbor Molecular Case Studies\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1101/mcs.a006193\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2022/2/1 0:00:00\",\"PubModel\":\"Print\",\"JCR\":\"Q3\",\"JCRName\":\"MEDICINE, RESEARCH & EXPERIMENTAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cold Spring Harbor Molecular Case Studies","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1101/mcs.a006193","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/2/1 0:00:00","PubModel":"Print","JCR":"Q3","JCRName":"MEDICINE, RESEARCH & EXPERIMENTAL","Score":null,"Total":0}
Ethylmalonic encephalopathy masquerading as meningococcemia.
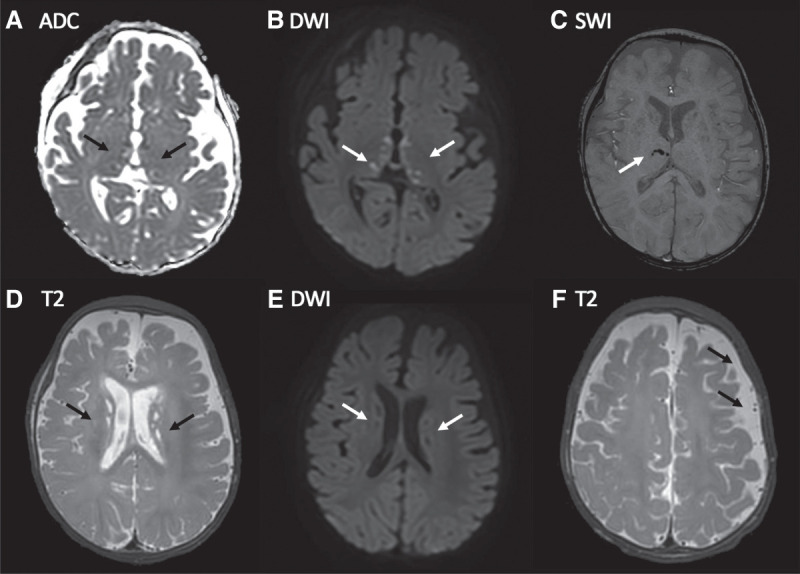
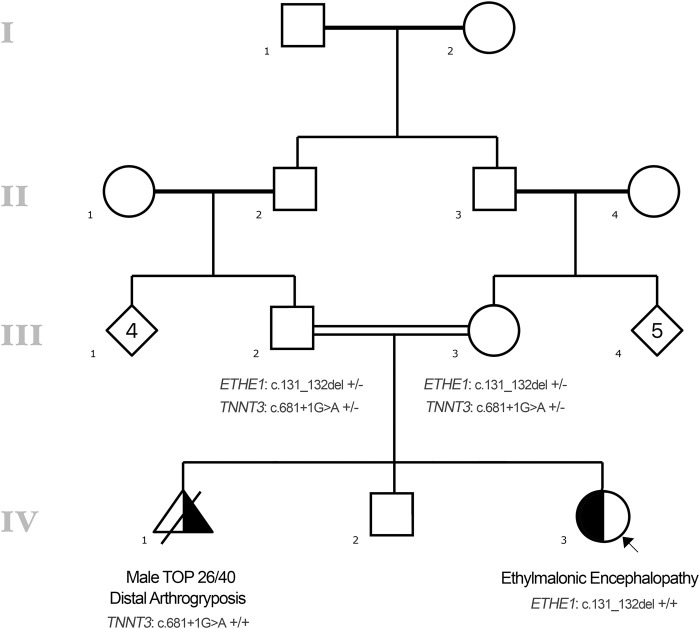
Ethylmalonic encephalopathy (MIM #602473) is a rare autosomal recessive metabolic condition caused by biallelic variants in ETHE1 (MIM #608451), characterized by global developmental delay, infantile hypotonia, seizures, and microvascular damage. The microvascular changes result in a pattern of relapsing spontaneous diffuse petechiae and purpura, positional acrocyanosis, and pedal edema, hemorrhagic suffusions of mucous membranes, and chronic diarrhea. Here, we describe an instructive case in which ethylmalonic encephalopathy masqueraded as meningococcal septicemia and shock. Ultrarapid whole-genome testing (time to result 60 h) and prompt biochemical analysis facilitated accurate diagnosis and counseling with rapid implementation of precision treatment for the metabolic crisis related to this condition. This case provides a timely reminder to consider rare genetic diagnoses when atypical features of more common conditions are present, with an early referral to ensure prompt biochemical and genomic diagnosis.
期刊介绍:
Cold Spring Harbor Molecular Case Studies is an open-access, peer-reviewed, international journal in the field of precision medicine. Articles in the journal present genomic and molecular analyses of individuals or cohorts alongside their clinical presentations and phenotypic information. The journal''s purpose is to rapidly share insights into disease development and treatment gained by application of genomics, proteomics, metabolomics, biomarker analysis, and other approaches. The journal covers the fields of cancer, complex diseases, monogenic disorders, neurological conditions, orphan diseases, infectious disease, gene therapy, and pharmacogenomics. It has a rapid peer-review process that is based on technical evaluation of the analyses performed, not the novelty of findings, and offers a swift, clear path to publication. The journal publishes: Research Reports presenting detailed case studies of individuals and small cohorts, Research Articles describing more extensive work using larger cohorts and/or functional analyses, Rapid Communications presenting the discovery of a novel variant and/or novel phenotype associated with a known disease gene, Rapid Cancer Communications presenting the discovery of a novel variant or combination of variants in a cancer type, Variant Discrepancy Resolution describing efforts to resolve differences or update variant interpretations in ClinVar through case-level data sharing, Follow-up Reports linked to previous observations, Plus Review Articles, Editorials, and Position Statements on best practices for research in precision medicine.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


