{"title":"基于分子对接的高通量虚拟筛选计算平台。","authors":"Baohua Zhang, Hui Li, Kunqian Yu, Zhong Jin","doi":"10.1007/s42514-021-00086-5","DOIUrl":null,"url":null,"abstract":"<p><p>Structure-based virtual screening is a key, routine computational method in computer-aided drug design. Such screening can be used to identify potentially highly active compounds, to speed up the progress of novel drug design. Molecular docking-based virtual screening can help find active compounds from large ligand databases by identifying the binding affinities between receptors and ligands. In this study, we analyzed the challenges of virtual screening, with the aim of identifying highly active compounds faster and more easily than is generally possible. We discuss the accuracy and speed of molecular docking software and the strategy of high-throughput molecular docking calculation, and we focus on current challenges and our solutions to these challenges of ultra-large-scale virtual screening. The development of Web services helps lower the barrier to drug virtual screening. We introduced some related web sites for docking and virtual screening, focusing on the development of pre- and post-processing interactive visualization and large-scale computing.</p>","PeriodicalId":29895,"journal":{"name":"CCF Transactions on High Performance Computing","volume":"4 1","pages":"63-74"},"PeriodicalIF":1.9000,"publicationDate":"2022-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8754542/pdf/","citationCount":"16","resultStr":"{\"title\":\"Molecular docking-based computational platform for high-throughput virtual screening.\",\"authors\":\"Baohua Zhang, Hui Li, Kunqian Yu, Zhong Jin\",\"doi\":\"10.1007/s42514-021-00086-5\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Structure-based virtual screening is a key, routine computational method in computer-aided drug design. Such screening can be used to identify potentially highly active compounds, to speed up the progress of novel drug design. Molecular docking-based virtual screening can help find active compounds from large ligand databases by identifying the binding affinities between receptors and ligands. In this study, we analyzed the challenges of virtual screening, with the aim of identifying highly active compounds faster and more easily than is generally possible. We discuss the accuracy and speed of molecular docking software and the strategy of high-throughput molecular docking calculation, and we focus on current challenges and our solutions to these challenges of ultra-large-scale virtual screening. The development of Web services helps lower the barrier to drug virtual screening. We introduced some related web sites for docking and virtual screening, focusing on the development of pre- and post-processing interactive visualization and large-scale computing.</p>\",\"PeriodicalId\":29895,\"journal\":{\"name\":\"CCF Transactions on High Performance Computing\",\"volume\":\"4 1\",\"pages\":\"63-74\"},\"PeriodicalIF\":1.9000,\"publicationDate\":\"2022-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8754542/pdf/\",\"citationCount\":\"16\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"CCF Transactions on High Performance Computing\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1007/s42514-021-00086-5\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2022/1/13 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q4\",\"JCRName\":\"COMPUTER SCIENCE, HARDWARE & ARCHITECTURE\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"CCF Transactions on High Performance Computing","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1007/s42514-021-00086-5","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/1/13 0:00:00","PubModel":"Epub","JCR":"Q4","JCRName":"COMPUTER SCIENCE, HARDWARE & ARCHITECTURE","Score":null,"Total":0}
Molecular docking-based computational platform for high-throughput virtual screening.
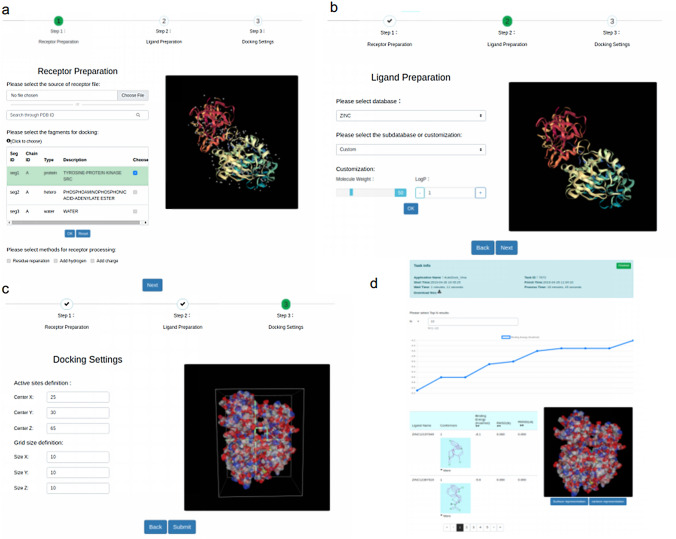
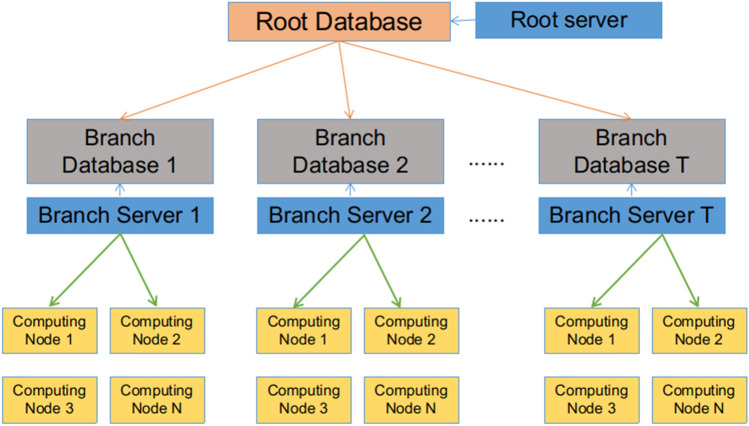
Structure-based virtual screening is a key, routine computational method in computer-aided drug design. Such screening can be used to identify potentially highly active compounds, to speed up the progress of novel drug design. Molecular docking-based virtual screening can help find active compounds from large ligand databases by identifying the binding affinities between receptors and ligands. In this study, we analyzed the challenges of virtual screening, with the aim of identifying highly active compounds faster and more easily than is generally possible. We discuss the accuracy and speed of molecular docking software and the strategy of high-throughput molecular docking calculation, and we focus on current challenges and our solutions to these challenges of ultra-large-scale virtual screening. The development of Web services helps lower the barrier to drug virtual screening. We introduced some related web sites for docking and virtual screening, focusing on the development of pre- and post-processing interactive visualization and large-scale computing.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


